

Structural basis of regulation and oligomerization of human cystathionine β-synthase, the central enzyme of transsulfuration
- PMID: 24043838
- PMCID: PMC3791738
- DOI: 10.1073/pnas.1313683110
Structural basis of regulation and oligomerization of human cystathionine β-synthase, the central enzyme of transsulfuration
Abstract
Cystathionine β-synthase (CBS) controls the flux of sulfur from methionine to cysteine, a precursor of glutathione, taurine, and H2S. CBS condenses serine and homocysteine to cystathionine with the help of three cofactors, heme, pyridoxal-5'-phosphate, and S-adenosyl-l-methionine. Inherited deficiency of CBS activity causes homocystinuria, the most frequent disorder of sulfur metabolism. We present the structure of the human enzyme, discuss the unique arrangement of the CBS domains in the C-terminal region, and propose how they interact with the catalytic core of the complementary subunit to regulate access to the catalytic site. This arrangement clearly contrasts with other proteins containing the CBS domain including the recent Drosophila melanogaster CBS structure. The absence of large conformational changes and the crystal structure of the partially activated pathogenic D444N mutant suggest that the rotation of CBS motifs and relaxation of loops delineating the entrance to the catalytic site represent the most likely molecular mechanism of CBS activation by S-adenosyl-l-methionine. Moreover, our data suggest how tetramers, the native quaternary structure of the mammalian CBS enzymes, are formed. Because of its central role in transsulfuration, redox status, and H2S biogenesis, CBS represents a very attractive therapeutic target. The availability of the structure will help us understand the pathogenicity of the numerous missense mutations causing inherited homocystinuria and will allow the rational design of compounds modulating CBS activity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
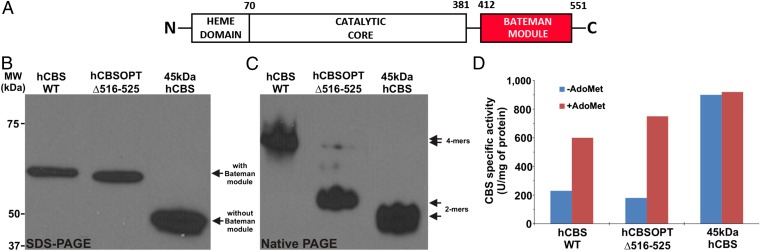
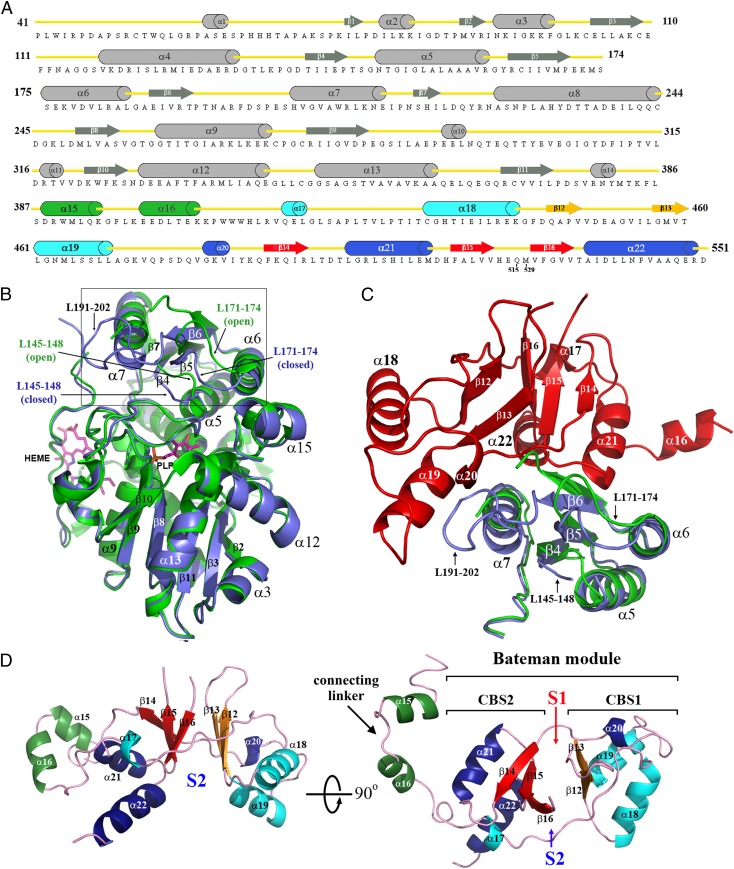
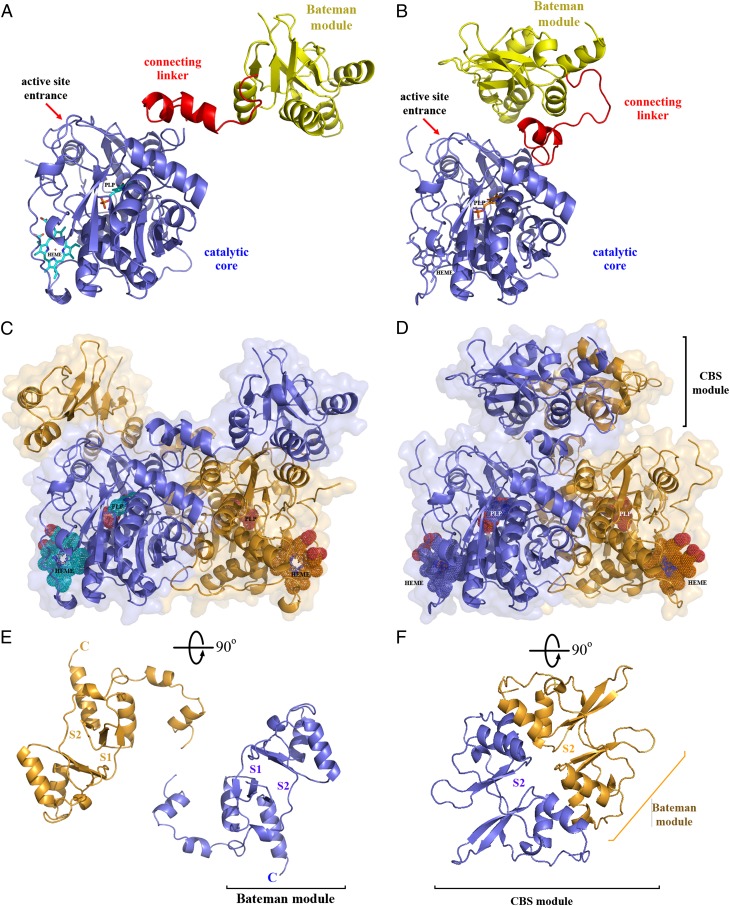
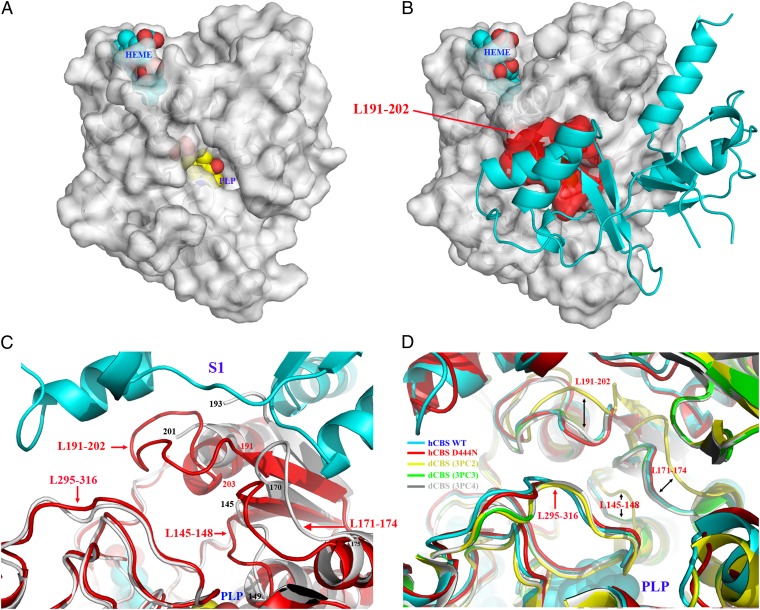
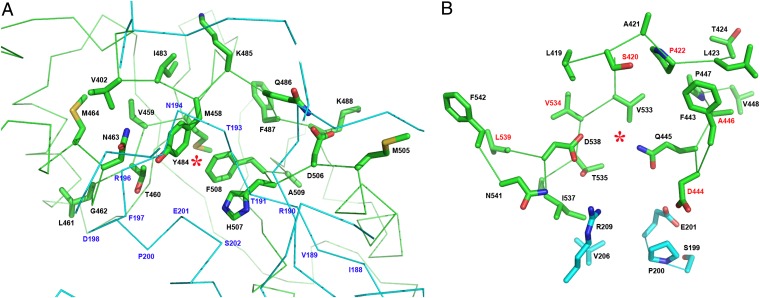
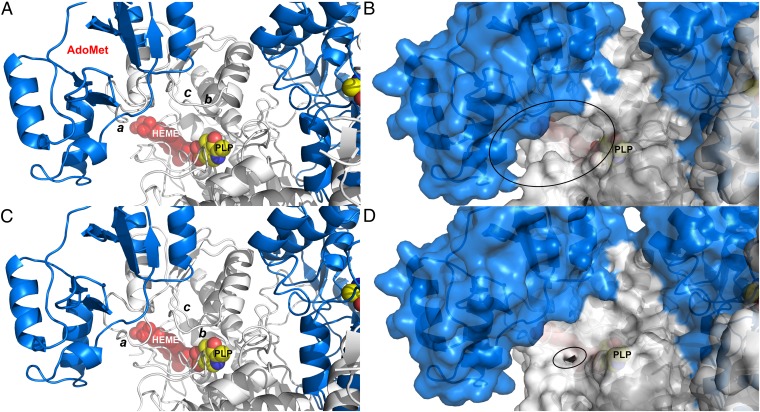
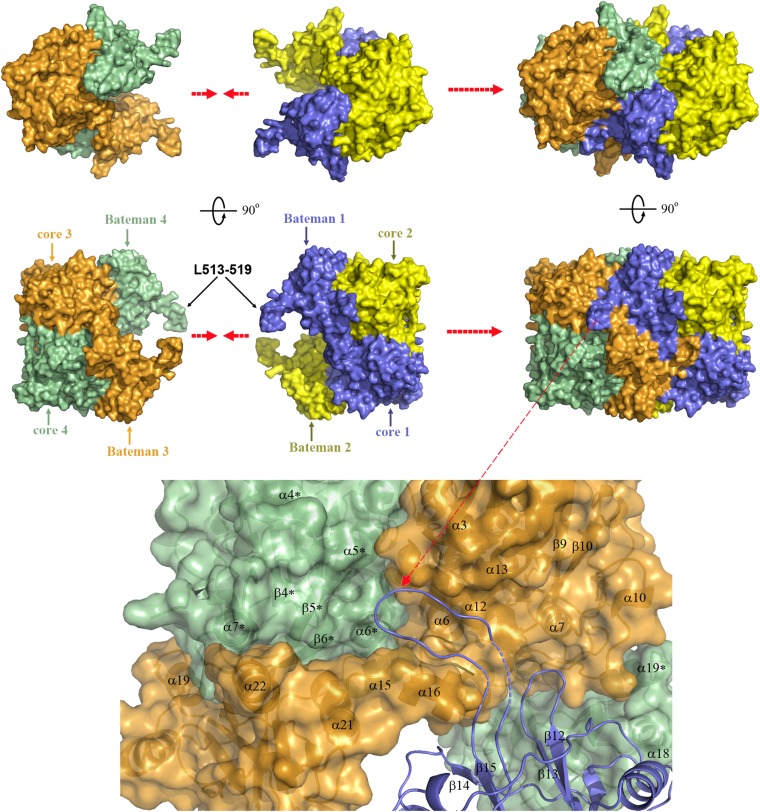
References
-
- Miles EW, Kraus JP. Cystathionine beta-synthase: Structure, function, regulation, and location of homocystinuria-causing mutations. J Biol Chem. 2004;279(29):29871–29874. - PubMed
-
- Castro R, Rivera I, Blom HJ, Jakobs C, Tavares de Almeida I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J Inherit Metab Dis. 2006;29(1):3–20. - PubMed
-
- Boers GHJ. Hyperhomocysteinemia as a risk factor for arterial and venous disease. A review of evidence and relevance. Thromb Haemost. 1997;78(1):520–522. - PubMed
-
- Makris M. Hyperhomocysteinemia is a risk factor for venous and arterial thrombosis. Br J Haematol. 1998;101(Suppl 1):18–20. - PubMed
-
- Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med. 1998;338(15):1042–1050. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
