

Genome-wide evidence for speciation with gene flow in Heliconius butterflies
- PMID: 24045163
- PMCID: PMC3814882
- DOI: 10.1101/gr.159426.113
Genome-wide evidence for speciation with gene flow in Heliconius butterflies
Abstract
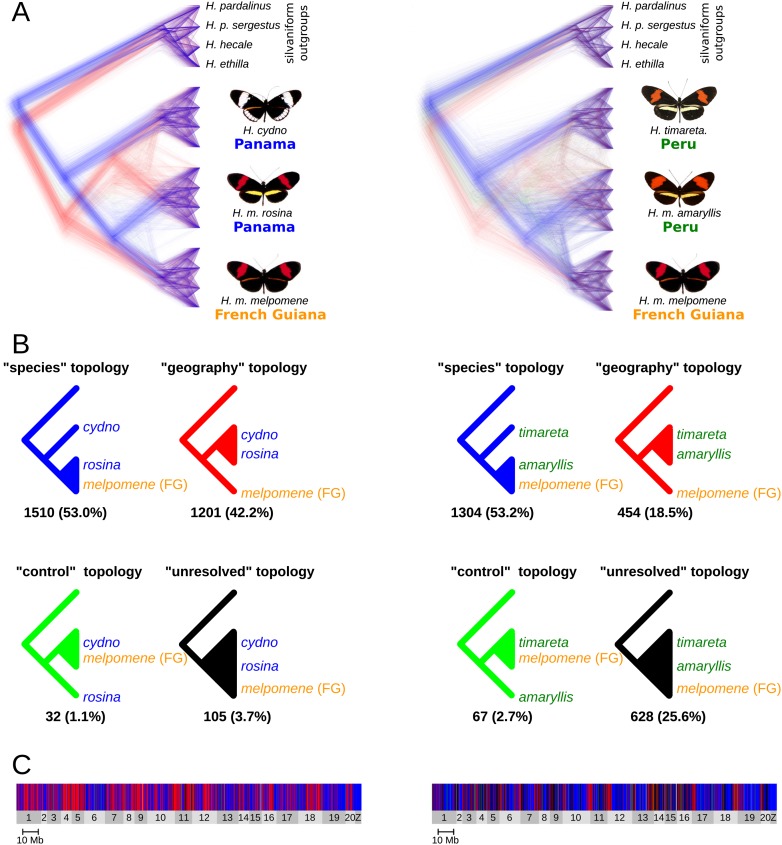
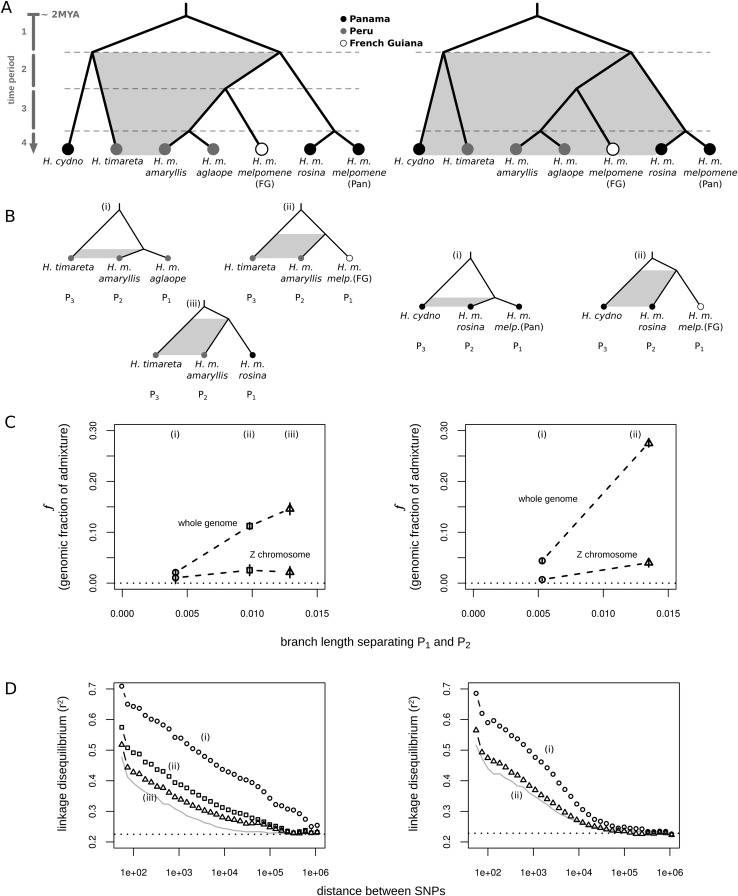
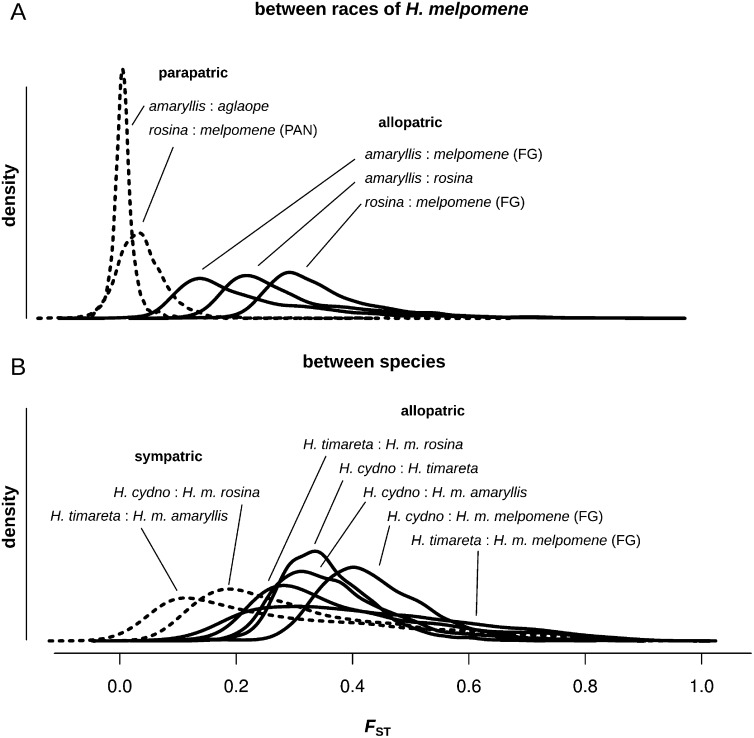
Most speciation events probably occur gradually, without complete and immediate reproductive isolation, but the full extent of gene flow between diverging species has rarely been characterized on a genome-wide scale. Documenting the extent and timing of admixture between diverging species can clarify the role of geographic isolation in speciation. Here we use new methodology to quantify admixture at different stages of divergence in Heliconius butterflies, based on whole-genome sequences of 31 individuals. Comparisons between sympatric and allopatric populations of H. melpomene, H. cydno, and H. timareta revealed a genome-wide trend of increased shared variation in sympatry, indicative of pervasive interspecific gene flow. Up to 40% of 100-kb genomic windows clustered by geography rather than by species, demonstrating that a very substantial fraction of the genome has been shared between sympatric species. Analyses of genetic variation shared over different time intervals suggested that admixture between these species has continued since early in speciation. Alleles shared between species during recent time intervals displayed higher levels of linkage disequilibrium than those shared over longer time intervals, suggesting that this admixture took place at multiple points during divergence and is probably ongoing. The signal of admixture was significantly reduced around loci controlling divergent wing patterns, as well as throughout the Z chromosome, consistent with strong selection for Müllerian mimicry and with known Z-linked hybrid incompatibility. Overall these results show that species divergence can occur in the face of persistent and genome-wide admixture over long periods of time.
Figures


Comment in
-
Evolution: Speciation goes with the flow.Nat Rev Genet. 2013 Nov;14(11):748. doi: 10.1038/nrg3597. Epub 2013 Oct 9. Nat Rev Genet. 2013. PMID: 24105276 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- BB/G008841/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/H014357/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/G006903/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- H01439X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 095831/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources