

Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer
- PMID: 24045185
- PMCID: PMC3838895
- DOI: 10.1158/1078-0432.CCR-12-3904
Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer
Abstract
Purpose: Amplification of MYC is one of the most common genetic alterations in lung cancer, contributing to a myriad of phenotypes associated with growth, invasion, and drug resistance. Murine genetics has established both the centrality of somatic alterations of Kras in lung cancer, as well as the dependency of mutant Kras tumors on MYC function. Unfortunately, drug-like small-molecule inhibitors of KRAS and MYC have yet to be realized. The recent discovery, in hematologic malignancies, that bromodomain and extra-terminal (BET) bromodomain inhibition impairs MYC expression and MYC transcriptional function established the rationale of targeting KRAS-driven non-small cell lung cancer (NSCLC) with BET inhibition.
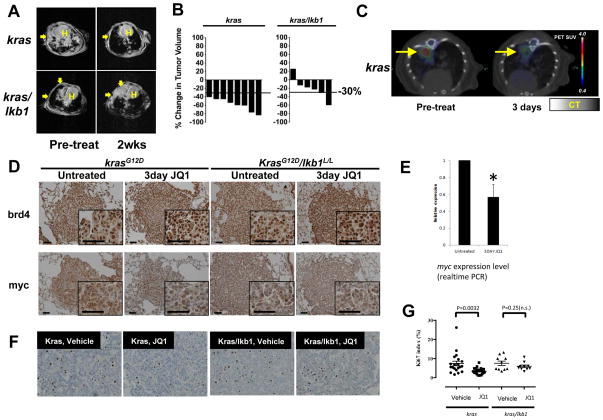
Experimental design: We performed functional assays to evaluate the effects of JQ1 in genetically defined NSCLC cell lines harboring KRAS and/or LKB1 mutations. Furthermore, we evaluated JQ1 in transgenic mouse lung cancer models expressing mutant kras or concurrent mutant kras and lkb1. Effects of bromodomain inhibition on transcriptional pathways were explored and validated by expression analysis.
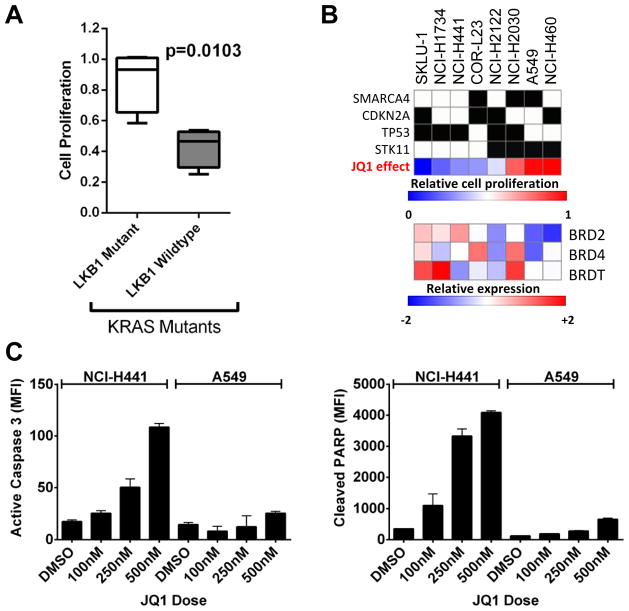
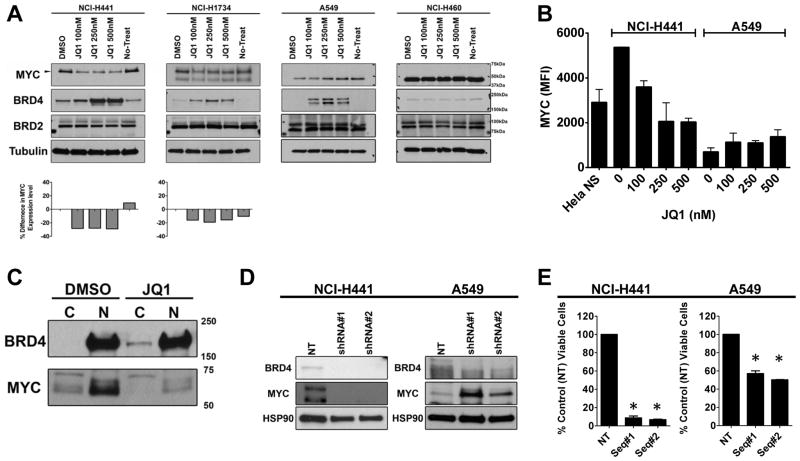
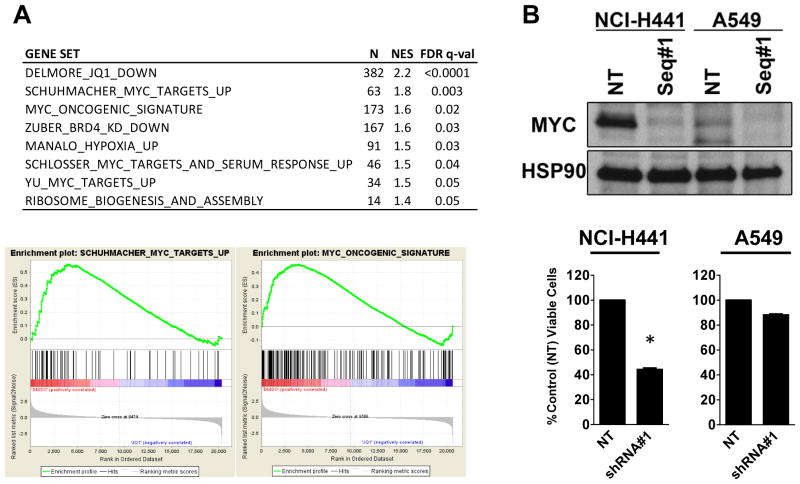
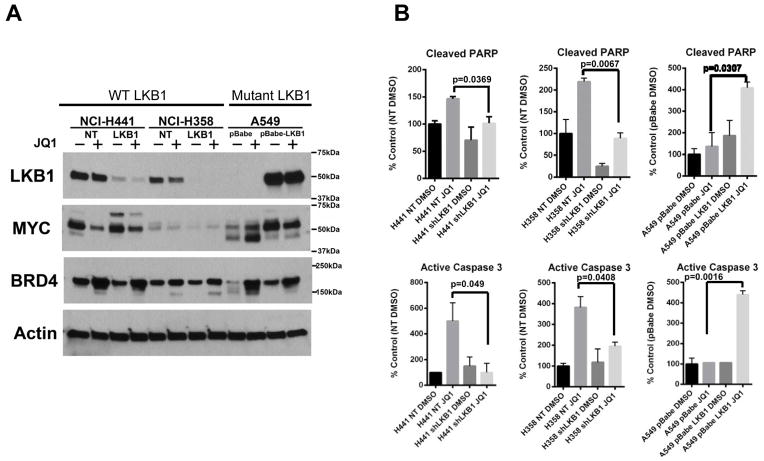
Results: Although JQ1 is broadly active in NSCLC cells, activity of JQ1 in mutant KRAS NSCLC is abrogated by concurrent alteration or genetic knockdown of LKB1. In sensitive NSCLC models, JQ1 treatment results in the coordinate downregulation of the MYC-dependent transcriptional program. We found that JQ1 treatment produces significant tumor regression in mutant kras mice. As predicted, tumors from mutant kras and lkb1 mice did not respond to JQ1.
Conclusion: Bromodomain inhibition comprises a promising therapeutic strategy for KRAS-mutant NSCLC with wild-type LKB1, via inhibition of MYC function. Clinical studies of BET bromodomain inhibitors in aggressive NSCLC will be actively pursued. Clin Cancer Res; 19(22); 6183-92. ©2013 AACR.
Conflict of interest statement
Drug-like BET bromodomain inhibitors created by Drs. Bradner and Qi have been licensed by the Dana-Farber Cancer Institute to Tensha Therapeutics (Cambridge, MA) for therapeutic development.
Drs. Bradner (Major) and Qi (Minor) consult for Tensha Therapeutics to contribute to the clinical translation of drug-like BET bromodomain inhibitors.
Dr. Kimmelman is a consultant for Forma Therapeutics (Minor).
Dr. Bradner and the Dana-Farber Cancer Institute have been allocated equity (minority) in Tensha Therapeutics.
Takeshi Shimamura, Zhao Chen, Margaret Soucheray, Julian Carretero, Eiki Kikuchi, Jeremy H. Tchaicha, Yandi Gao, Katherine A. Cheng, Travis J. Cohoon, Esra Akbay, Andrew L. Kung and Kwok-Kin Wong
No conflicts of interest
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- RC2 CA147940/CA/NCI NIH HHS/United States
- P50CA090578/CA/NCI NIH HHS/United States
- R01 CA137008/CA/NCI NIH HHS/United States
- R01 CA122794/CA/NCI NIH HHS/United States
- R01 CA137181/CA/NCI NIH HHS/United States
- 1U01CA141576/CA/NCI NIH HHS/United States
- CA137008/CA/NCI NIH HHS/United States
- P50 CA090578/CA/NCI NIH HHS/United States
- CA147940/CA/NCI NIH HHS/United States
- U01 CA141576/CA/NCI NIH HHS/United States
- P01 CA120964/CA/NCI NIH HHS/United States
- CA137181/CA/NCI NIH HHS/United States
- P01 CA154303/CA/NCI NIH HHS/United States
- R01 CA163896/CA/NCI NIH HHS/United States
- R01 CA166480/CA/NCI NIH HHS/United States
- R01 CA157490/CA/NCI NIH HHS/United States
- CA137008-01/CA/NCI NIH HHS/United States
- CA122794/CA/NCI NIH HHS/United States
- R01 CA140594/CA/NCI NIH HHS/United States
- CA140594/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
