

A novel intermediate mucolipidosis II/IIIαβ caused by GNPTAB mutation in the cytosolic N-terminal domain
- PMID: 24045841
- PMCID: PMC3992569
- DOI: 10.1038/ejhg.2013.207
A novel intermediate mucolipidosis II/IIIαβ caused by GNPTAB mutation in the cytosolic N-terminal domain
Abstract
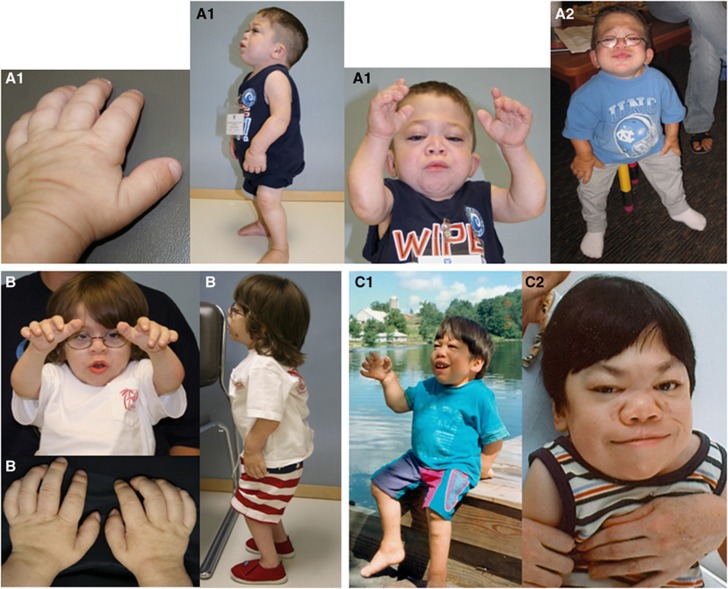
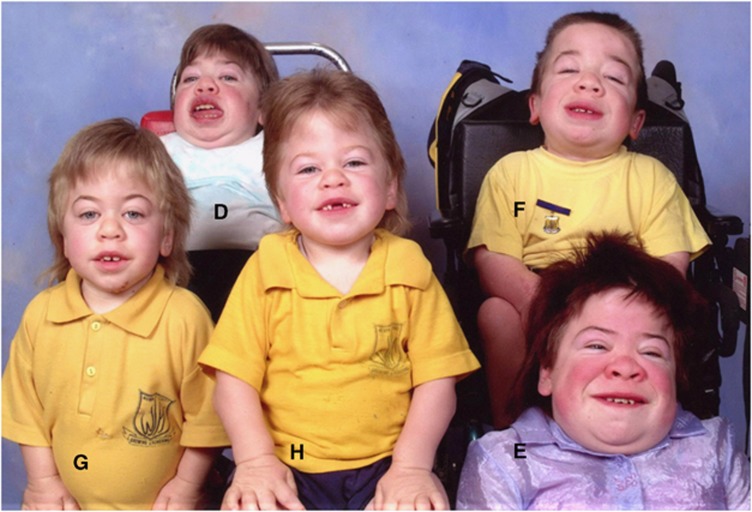
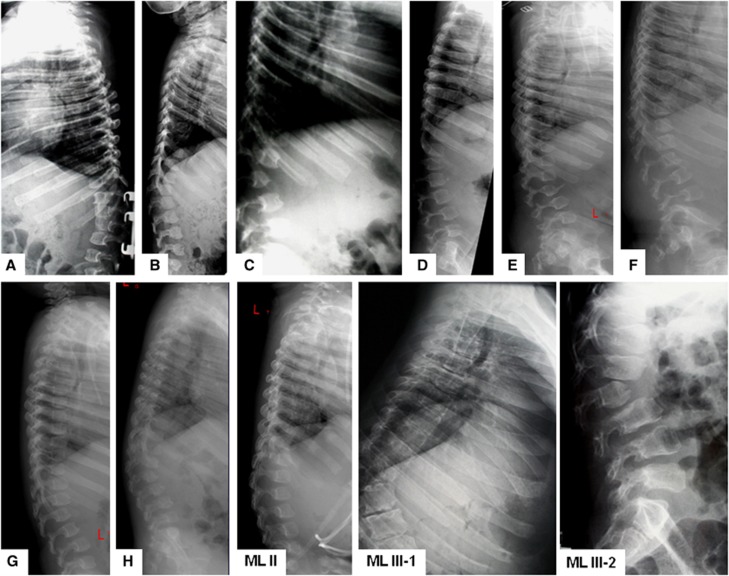
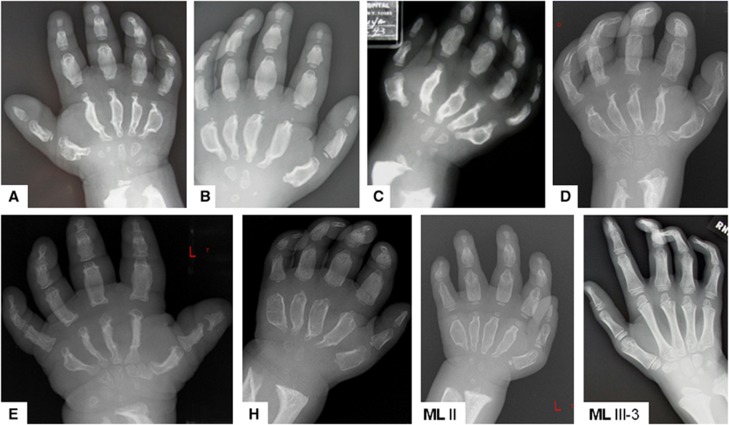
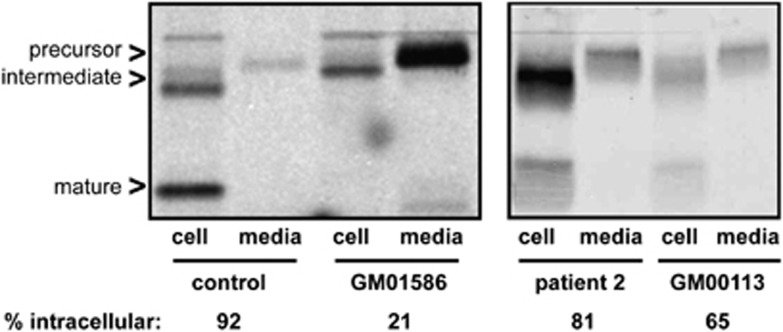
Mucolipidosis (ML) II and ML IIIα/β are allelic autosomal recessive metabolic disorders due to mutations in GNPTAB. The gene encodes the enzyme UDP-GlcNAc-1-phosphotransferase (GNPT), which is critical to proper trafficking of lysosomal acid hydrolases. The ML phenotypic spectrum is dichotomous. Criteria set for defining ML II and ML IIIα/β are inclusive for all but the few patients with phenotypes that span the archetypes. Clinical and biochemical findings of the 'intermediate' ML in eight patients with the c.10A>C missense mutation in GNPTAB are presented to define this intermediate ML and provide a broader insight into ML pathogenesis. Extensive clinical information, including radiographic examinations at various ages, was obtained from a detailed study of all patients. GNPTAB was sequenced in probands and parents. GNPT activity was measured and cathepsin D sorting assays were performed in fibroblasts. Intermediate ML patients who share the c.10A>C/p.K4Q mutation in GNPTAB demonstrate a distinct, consistent phenotype similar to ML II in physical and radiographic features and to ML IIIα/β in psychomotor development and life expectancy. GNPT activity is reduced to 7-12% but the majority of newly synthesized cathepsin D remains intracellular. The GNPTAB c.10A>C/p.K4Q missense allele results in an intermediate ML II/III with distinct clinical and biochemical characteristics. This delineation strengthens the utility of the discontinuous genotype-phenotype correlation in ML II and ML IIIα/β and prompts additional studies on the tissue-specific pathogenesis in GNPT-deficient ML.
Figures
References
-
- Kobayashi H, Takahashi-Fujigasaki J, Takahiro F, et al. Pathology of the first autopsy case diagnosed as mucolipidosis type III α/β suggesting autophagic dysfunction. Mol Genet Metab. 2011;102:170–175. - PubMed
-
- Kozlowski K, Lipson A, Carey W. Mild I-cell disease, or severe pseudo-Hurler polydystrophy in three siblings; further evidence for intermediate forms of mucolipidosis II and III. Radiological features. Radiol Med. 1991;82:847–851. - PubMed
-
- Thomas GH, Taylor HA, Reynolds LW, et al. Mucolipidosis 3 (Pseudo-Hurler polydystrophy): multiple lysosomal enzyme abnormalities in serum and cultured fibroblast cells. Pediatr Res. 1973;7:751–756. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
