

eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases
- PMID: 24046447
- PMCID: PMC3860306
- DOI: 10.1242/jcs.115972
eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases
Erratum in
- J Cell Sci. 2014 May 1;127(Pt 9):2120
Abstract
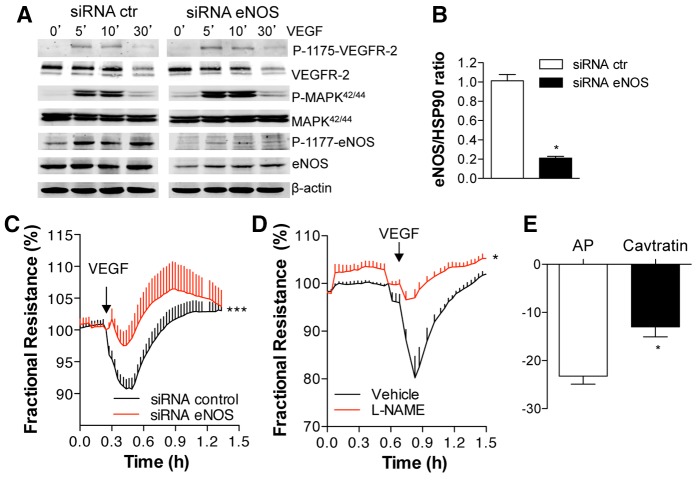
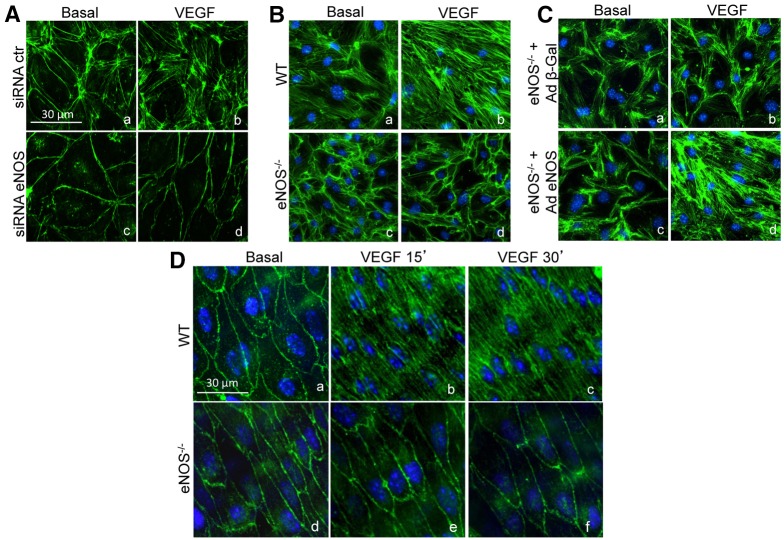
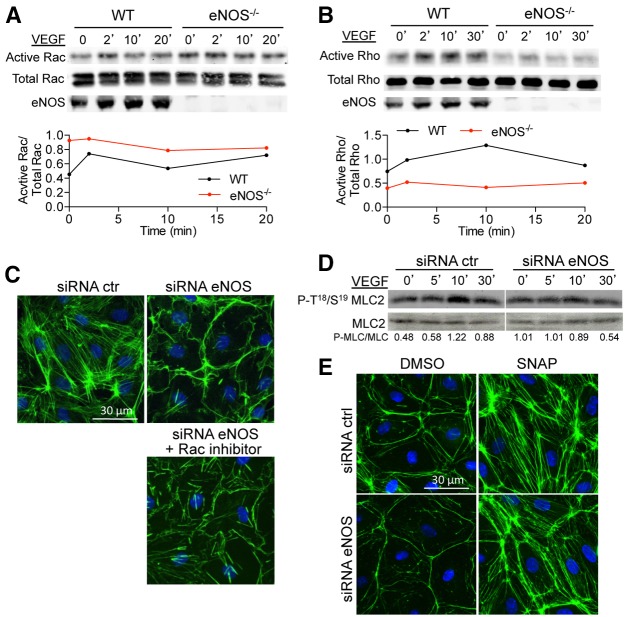
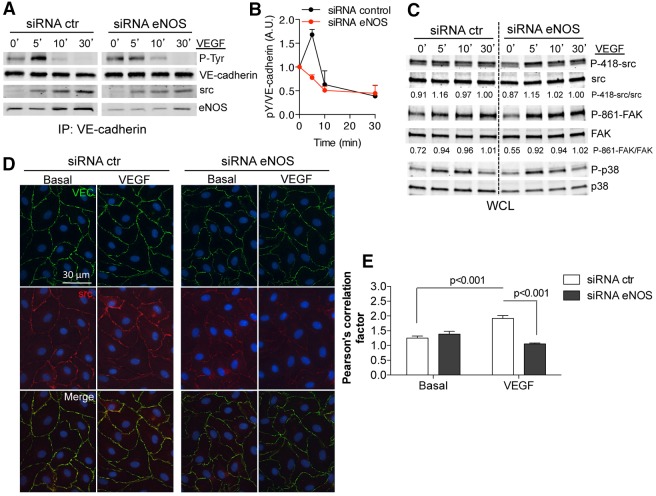
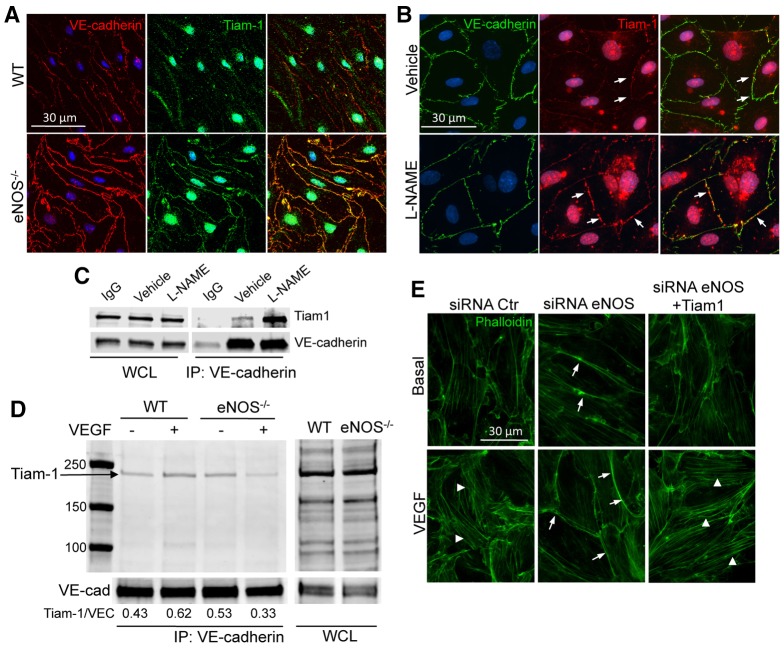
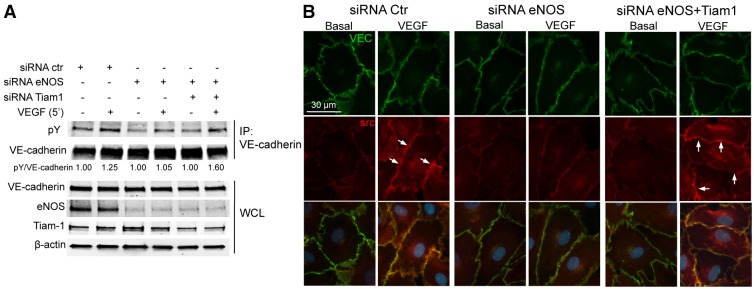
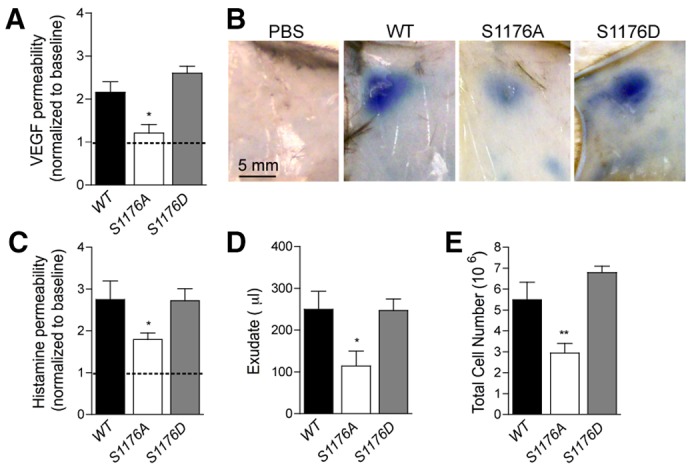
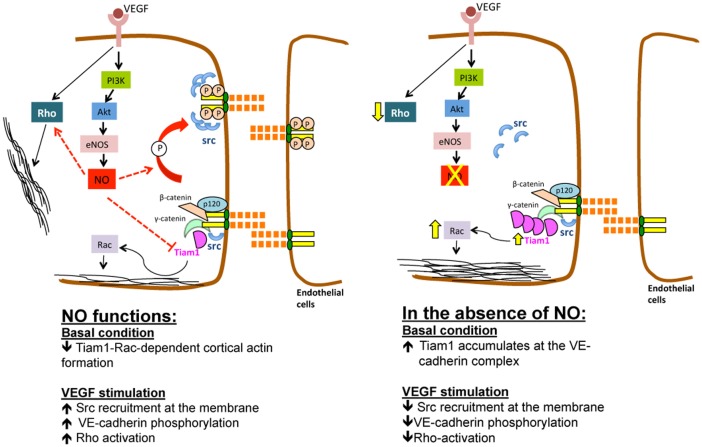
Transient disruption of endothelial adherens junctions and cytoskeletal remodeling are responsible for increases in vascular permeability induced by inflammatory stimuli and vascular endothelial growth factor (VEGF). Nitric oxide (NO) produced by endothelial NO synthase (eNOS) is crucial for VEGF-induced changes in permeability in vivo; however, the molecular mechanism by which endogenous NO modulates endothelial permeability is not clear. Here, we show that the lack of eNOS reduces VEGF-induced permeability, an effect mediated by enhanced activation of the Rac GTPase and stabilization of cortical actin. The loss of NO increased the recruitment of the Rac guanine-nucleotide-exchange factor (GEF) TIAM1 to adherens junctions and VE-cadherin (also known as cadherin 5), and reduced Rho activation and stress fiber formation. In addition, NO deficiency reduced VEGF-induced VE-cadherin phosphorylation and impaired the localization, but not the activation, of c-Src to cell junctions. The physiological role of eNOS activation is clear given that VEGF-, histamine- and inflammation-induced vascular permeability is reduced in mice bearing a non-phosphorylatable knock-in mutation of the key eNOS phosphorylation site S1176. Thus, NO is crucial for Rho GTPase-dependent regulation of cytoskeletal architecture leading to reversible changes in vascular permeability.
Keywords: Cadherin 5; Cytoskeleton; Nitric oxide; Src; VE-cadherin; VEGF; eNOS.
Figures
References
-
- Angelini D. J., Hyun S. W., Grigoryev D. N., Garg P., Gong P., Singh I. S., Passaniti A., Hasday J. D., Goldblum S. E. (2006). TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. 291, L1232–L1245 10.1152/ajplung.00109.2006 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
