

Draft genome sequence of the mulberry tree Morus notabilis
- PMID: 24048436
- PMCID: PMC3791463
- DOI: 10.1038/ncomms3445
Draft genome sequence of the mulberry tree Morus notabilis
Abstract
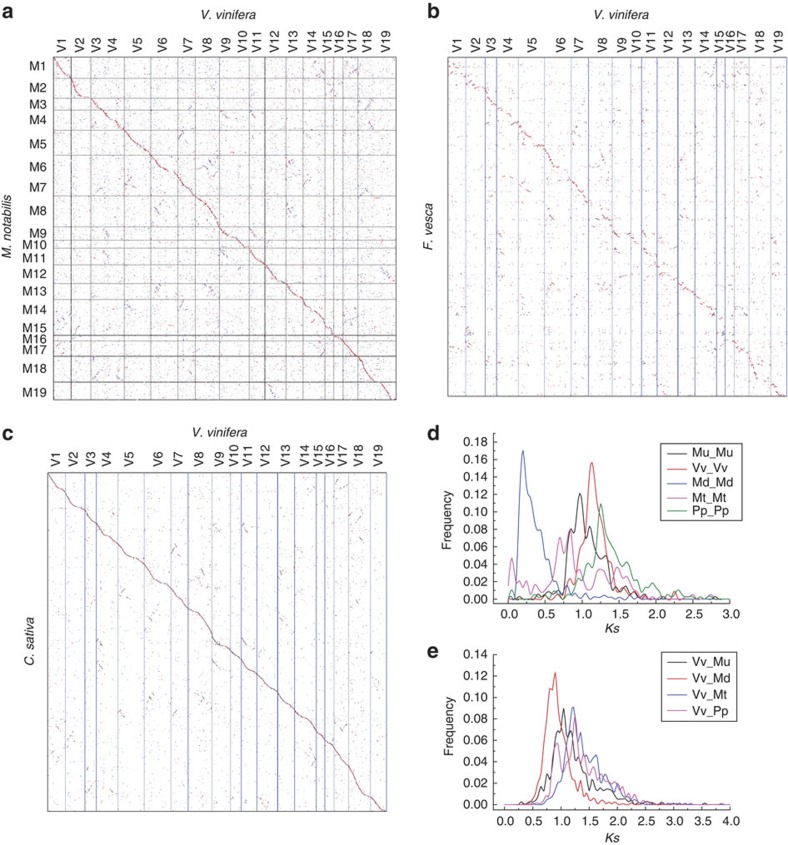
Human utilization of the mulberry-silkworm interaction started at least 5,000 years ago and greatly influenced world history through the Silk Road. Complementing the silkworm genome sequence, here we describe the genome of a mulberry species Morus notabilis. In the 330-Mb genome assembly, we identify 128 Mb of repetitive sequences and 29,338 genes, 60.8% of which are supported by transcriptome sequencing. Mulberry gene sequences appear to evolve ~3 times faster than other Rosales, perhaps facilitating the species' spread worldwide. The mulberry tree is among a few eudicots but several Rosales that have not preserved genome duplications in more than 100 million years; however, a neopolyploid series found in the mulberry tree and several others suggest that new duplications may confer benefits. Five predicted mulberry miRNAs are found in the haemolymph and silk glands of the silkworm, suggesting interactions at molecular levels in the plant-herbivore relationship. The identification and analyses of mulberry genes involved in diversifying selection, resistance and protease inhibitor expressed in the laticifers will accelerate the improvement of mulberry plants.
Figures
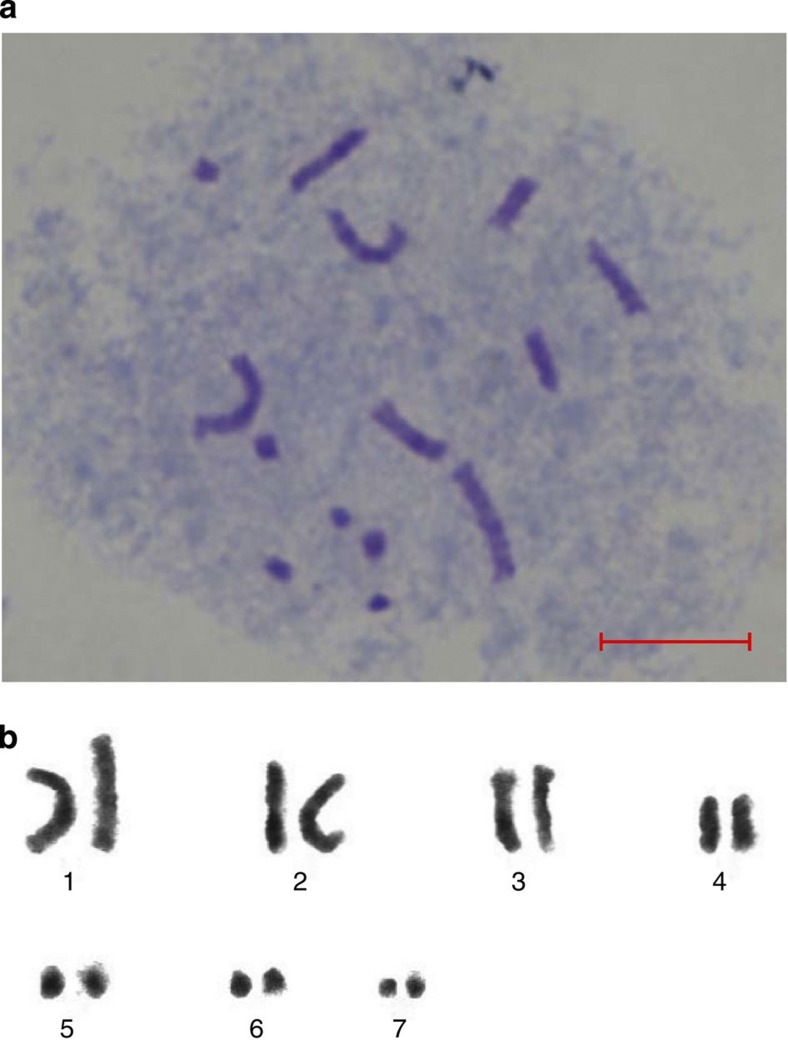

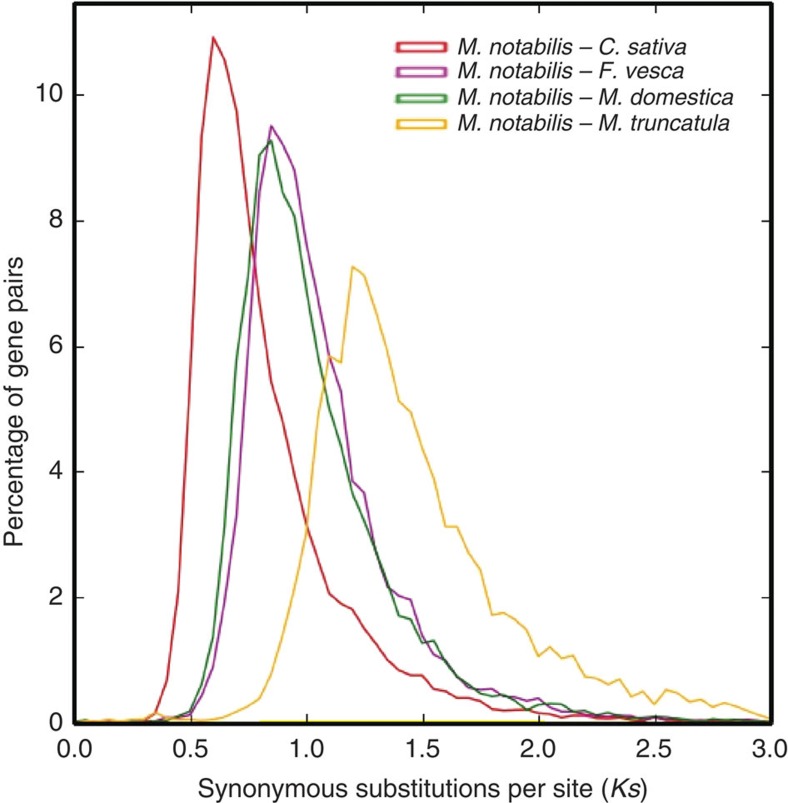
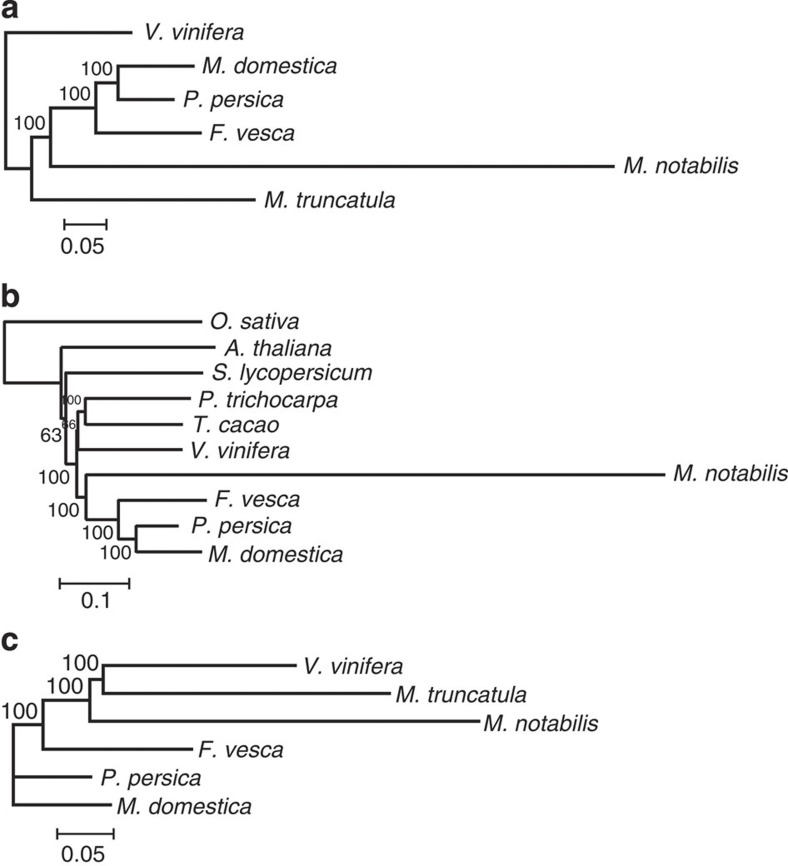
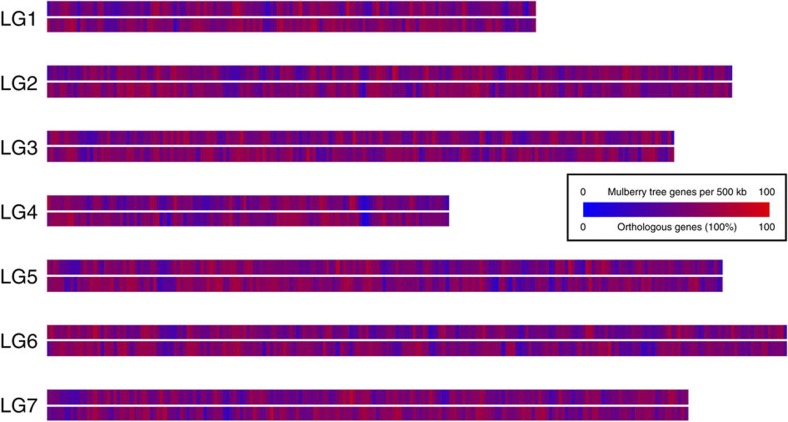
References
-
- Barber E. J. W. Prehistoric Textiles: The Development Of Cloth In The Neolithic And Bronze Ages With Special Reference To The Aegean Princeton University Press (1991).
-
- Clement W. L. & Weiblen G. D. Morphological evolution in the mulberry family (Moraceae). Syst. Bot. 34, 530–552 (2009).
-
- Nepal M. P. & Ferguson C. J. Phylogenetics of Morus (Moraceae) inferred from ITS and trnL-trnF sequence data. Syst. Bot. 37, 442–450 (2012).
-
- Sánchez M. D. World distribution and utilization of mulberry, potential for animal feeding. FAO Electron. Conf. Mulberry Animal Prod. (Morus1-L) 1–11 (2000).
-
- Kim S. Y. et al. Antioxidative flavonoids from the leaves of Morus alba. Arch. Pharm. Res. 22, 81–85 (1999). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
