

DEXUS: identifying differential expression in RNA-Seq studies with unknown conditions
- PMID: 24049071
- PMCID: PMC3834838
- DOI: 10.1093/nar/gkt834
DEXUS: identifying differential expression in RNA-Seq studies with unknown conditions
Abstract
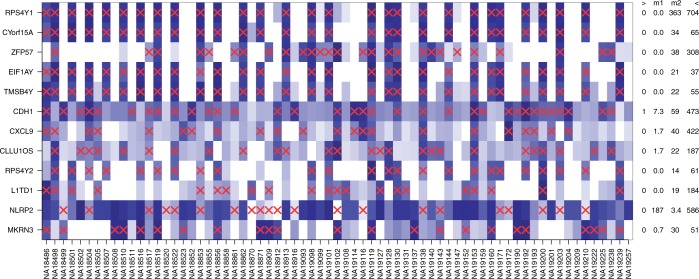
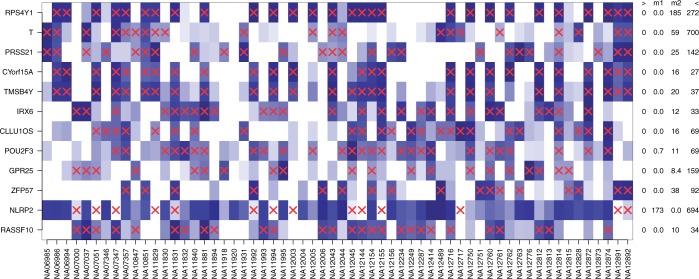
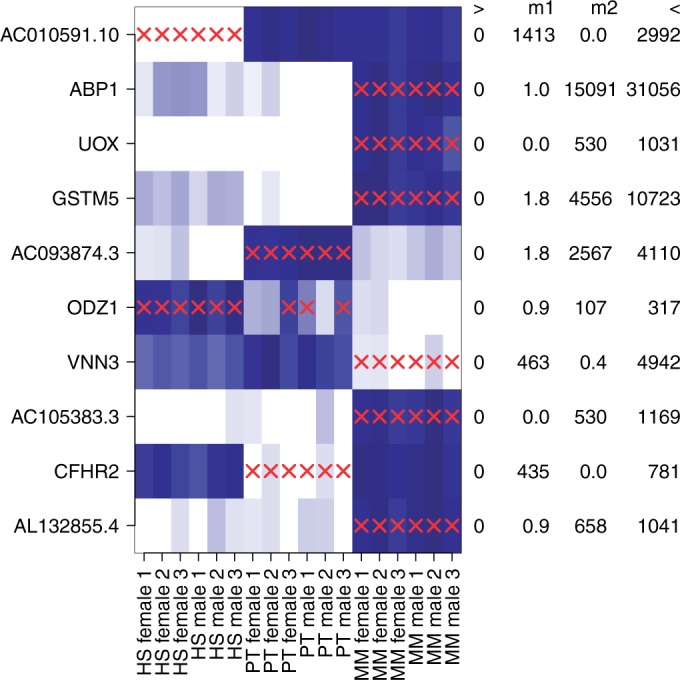
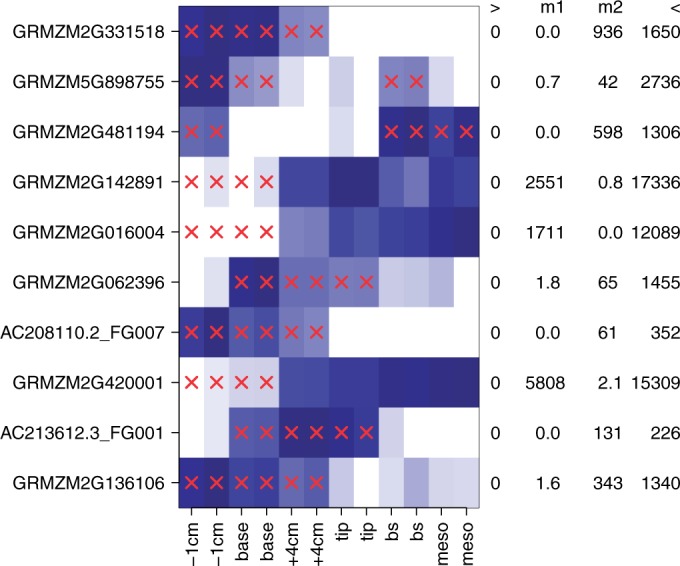
Detection of differential expression in RNA-Seq data is currently limited to studies in which two or more sample conditions are known a priori. However, these biological conditions are typically unknown in cohort, cross-sectional and nonrandomized controlled studies such as the HapMap, the ENCODE or the 1000 Genomes project. We present DEXUS for detecting differential expression in RNA-Seq data for which the sample conditions are unknown. DEXUS models read counts as a finite mixture of negative binomial distributions in which each mixture component corresponds to a condition. A transcript is considered differentially expressed if modeling of its read counts requires more than one condition. DEXUS decomposes read count variation into variation due to noise and variation due to differential expression. Evidence of differential expression is measured by the informative/noninformative (I/NI) value, which allows differentially expressed transcripts to be extracted at a desired specificity (significance level) or sensitivity (power). DEXUS performed excellently in identifying differentially expressed transcripts in data with unknown conditions. On 2400 simulated data sets, I/NI value thresholds of 0.025, 0.05 and 0.1 yielded average specificities of 92, 97 and 99% at sensitivities of 76, 61 and 38%, respectively. On real-world data sets, DEXUS was able to detect differentially expressed transcripts related to sex, species, tissue, structural variants or quantitative trait loci. The DEXUS R package is publicly available from Bioconductor and the scripts for all experiments are available at http://www.bioinf.jku.at/software/dexus/.
Figures
References
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods. 2008;5:621–628. - PubMed
-
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
