

Renal β-intercalated cells maintain body fluid and electrolyte balance
- PMID: 24051376
- PMCID: PMC3784520
- DOI: 10.1172/JCI63492
Renal β-intercalated cells maintain body fluid and electrolyte balance
Abstract
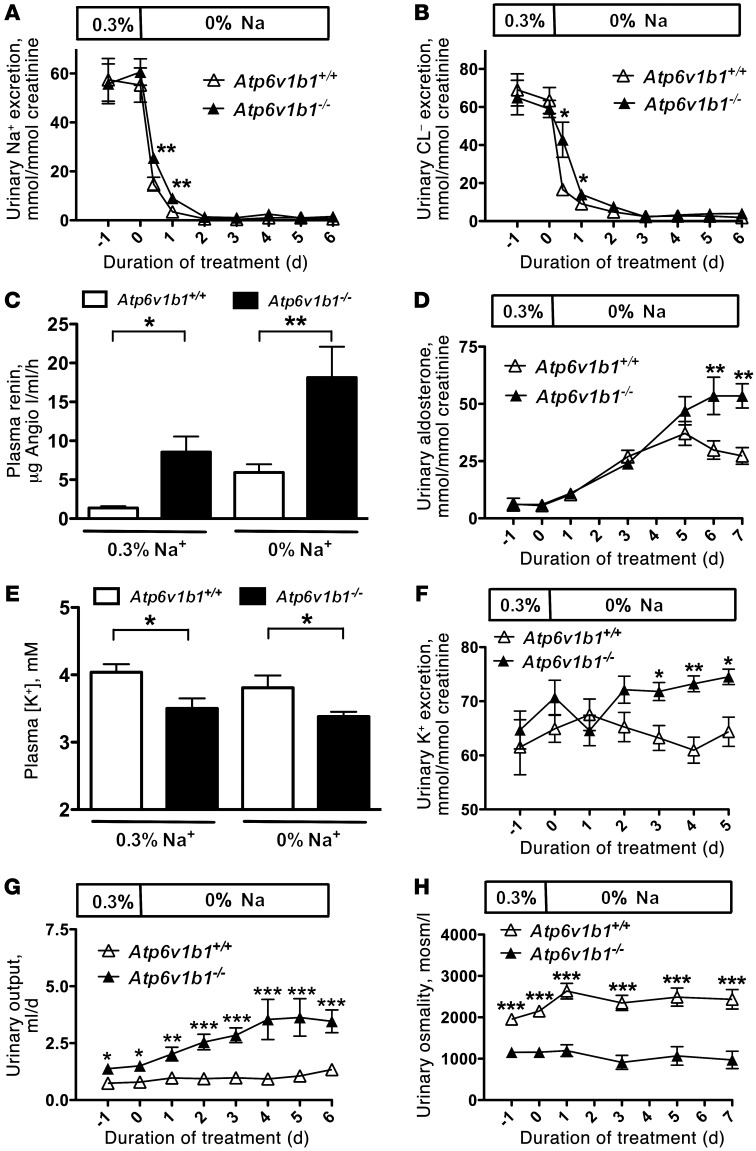
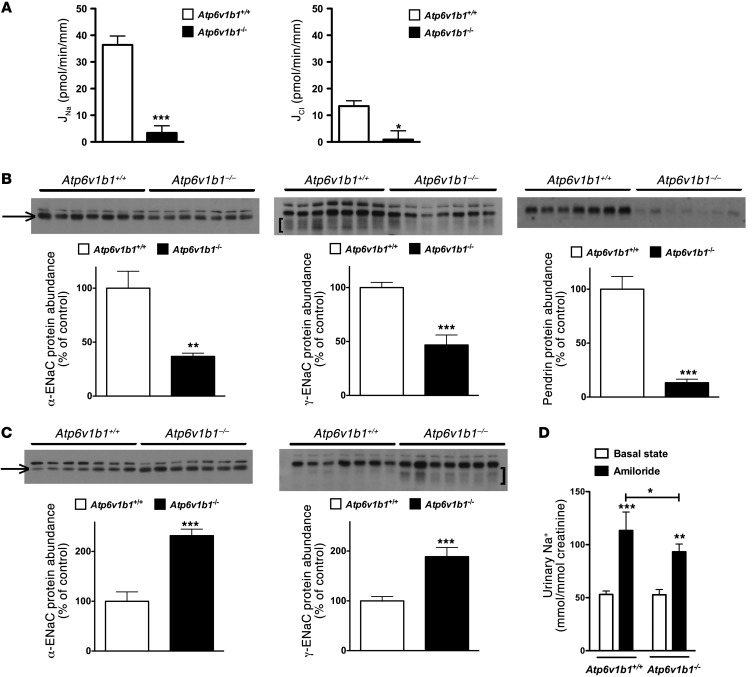
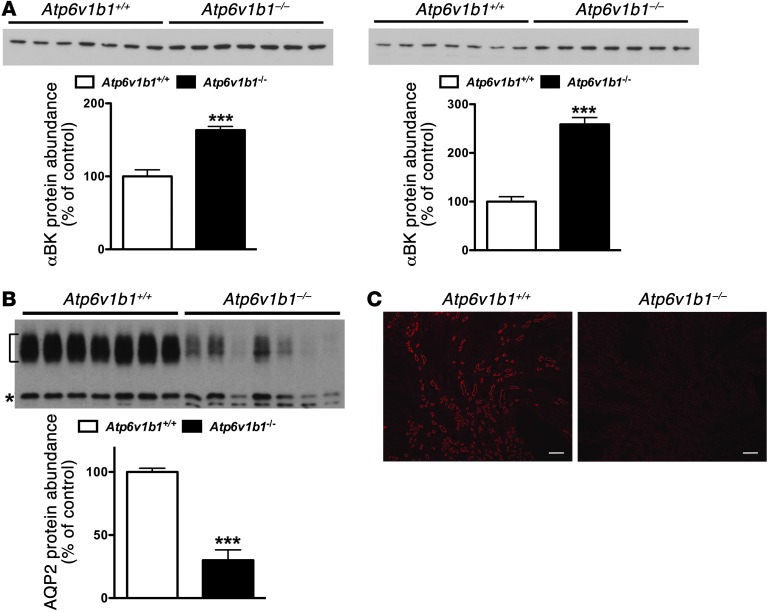
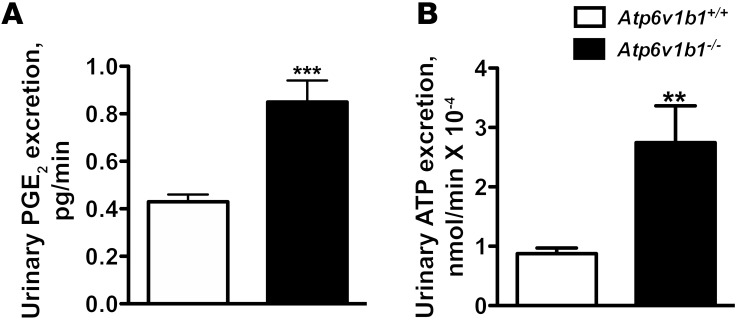
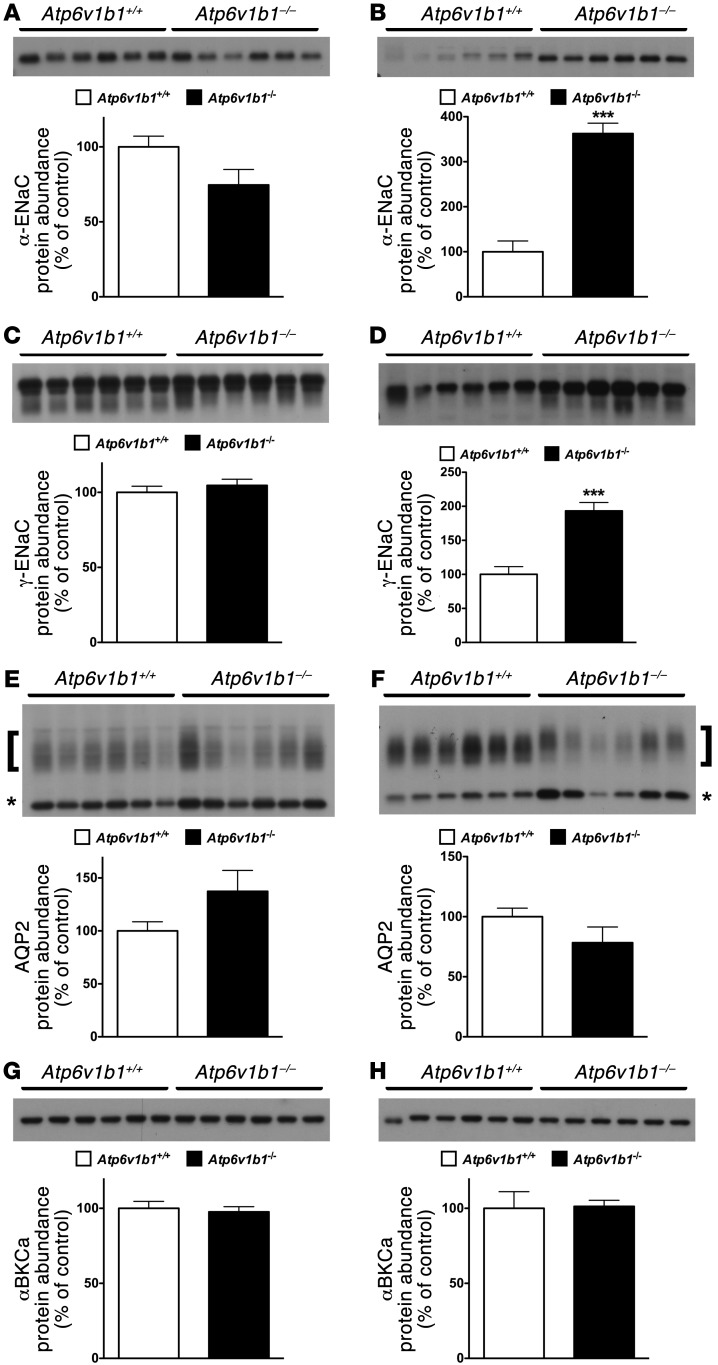
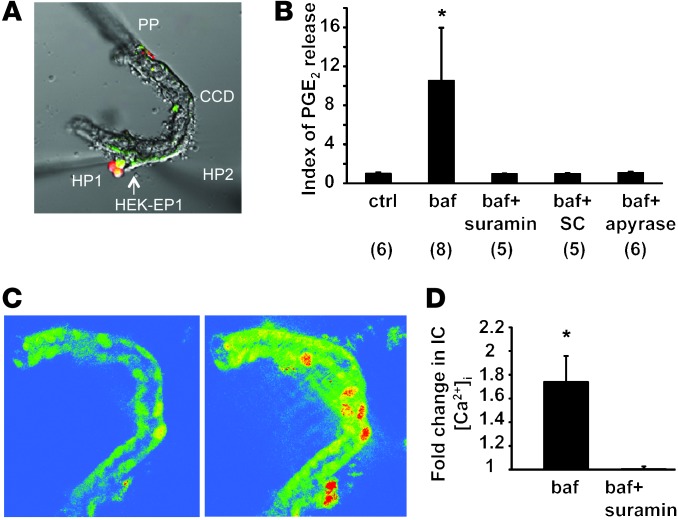
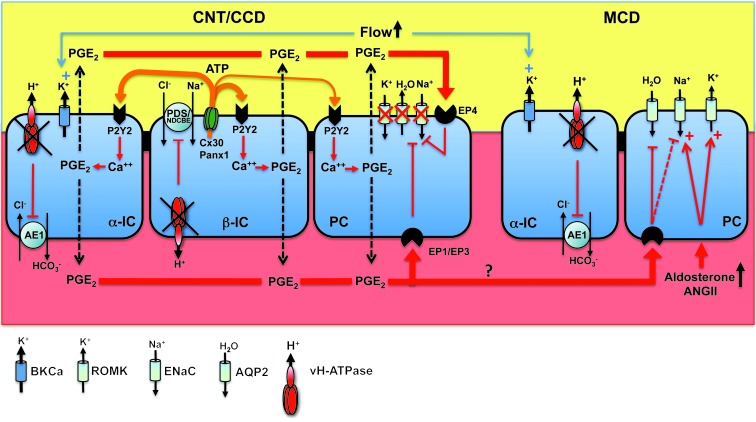
Inactivation of the B1 proton pump subunit (ATP6V1B1) in intercalated cells (ICs) leads to type I distal renal tubular acidosis (dRTA), a disease associated with salt- and potassium-losing nephropathy. Here we show that mice deficient in ATP6V1B1 (Atp6v1b1-/- mice) displayed renal loss of NaCl, K+, and water, causing hypovolemia, hypokalemia, and polyuria. We demonstrated that NaCl loss originated from the cortical collecting duct, where activity of both the epithelial sodium channel (ENaC) and the pendrin/Na(+)-driven chloride/bicarbonate exchanger (pendrin/NDCBE) transport system was impaired. ENaC was appropriately increased in the medullary collecting duct, suggesting a localized inhibition in the cortex. We detected high urinary prostaglandin E2 (PGE2) and ATP levels in Atp6v1b1-/- mice. Inhibition of PGE2 synthesis in vivo restored ENaC protein levels specifically in the cortex. It also normalized protein levels of the large conductance calcium-activated potassium channel and the water channel aquaporin 2, and improved polyuria and hypokalemia in mutant mice. Furthermore, pharmacological inactivation of the proton pump in β-ICs induced release of PGE2 through activation of calcium-coupled purinergic receptors. In the present study, we identified ATP-triggered PGE2 paracrine signaling originating from β-ICs as a mechanism in the development of the hydroelectrolytic imbalance associated with dRTA. Our data indicate that in addition to principal cells, ICs are also critical in maintaining sodium balance and, hence, normal vascular volume and blood pressure.
Figures
Comment in
-
Opening lines of communication in the distal nephron.J Clin Invest. 2013 Oct;123(10):4139-41. doi: 10.1172/JCI71944. Epub 2013 Sep 24. J Clin Invest. 2013. PMID: 24051382 Free PMC article.
-
Basic research: Salt wasting in distal renal tubular acidosis-new look, old problem.Nat Rev Nephrol. 2013 Dec;9(12):712-3. doi: 10.1038/nrneph.2013.235. Epub 2013 Nov 5. Nat Rev Nephrol. 2013. PMID: 24189652
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
