

Recent human adaptation: genomic approaches, interpretation and insights
- PMID: 24052086
- PMCID: PMC4882605
- DOI: 10.1038/nrg3604
Recent human adaptation: genomic approaches, interpretation and insights
Abstract
The recent availability of genomic data has spurred many genome-wide studies of human adaptation in different populations worldwide. Such studies have provided insights into novel candidate genes and pathways that are putatively involved in adaptation to different environments, diets and disease prevalence. However, much work is needed to translate these results into candidate adaptive variants that are biologically interpretable. In this Review, we discuss methods that may help to identify true biological signals of selection and studies that incorporate complementary phenotypic and functional data. We conclude with recommendations for future studies that focus on opportunities to use integrative genomics methodologies in human adaptation studies.
Figures
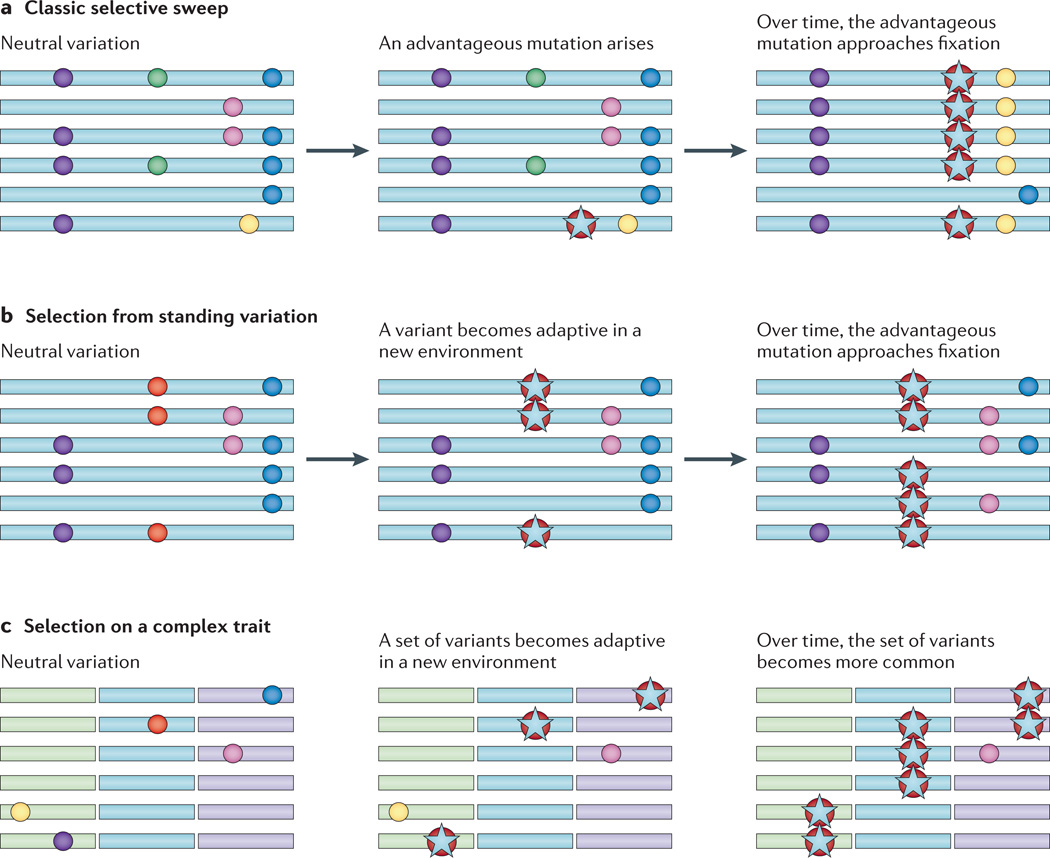
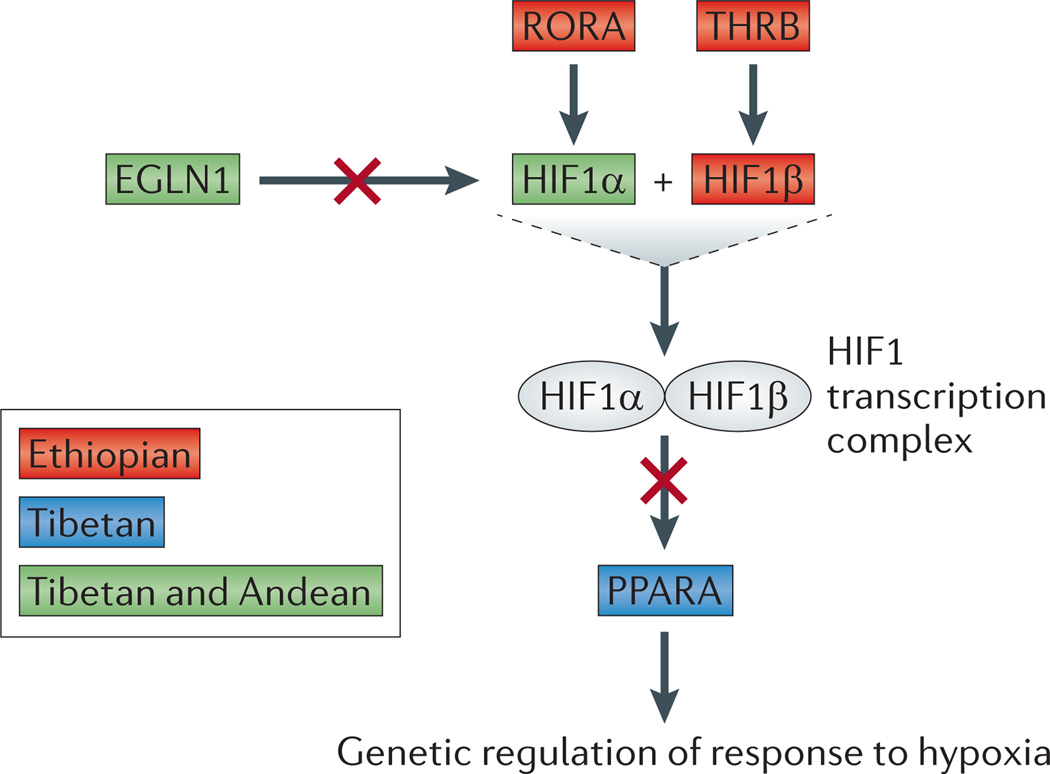
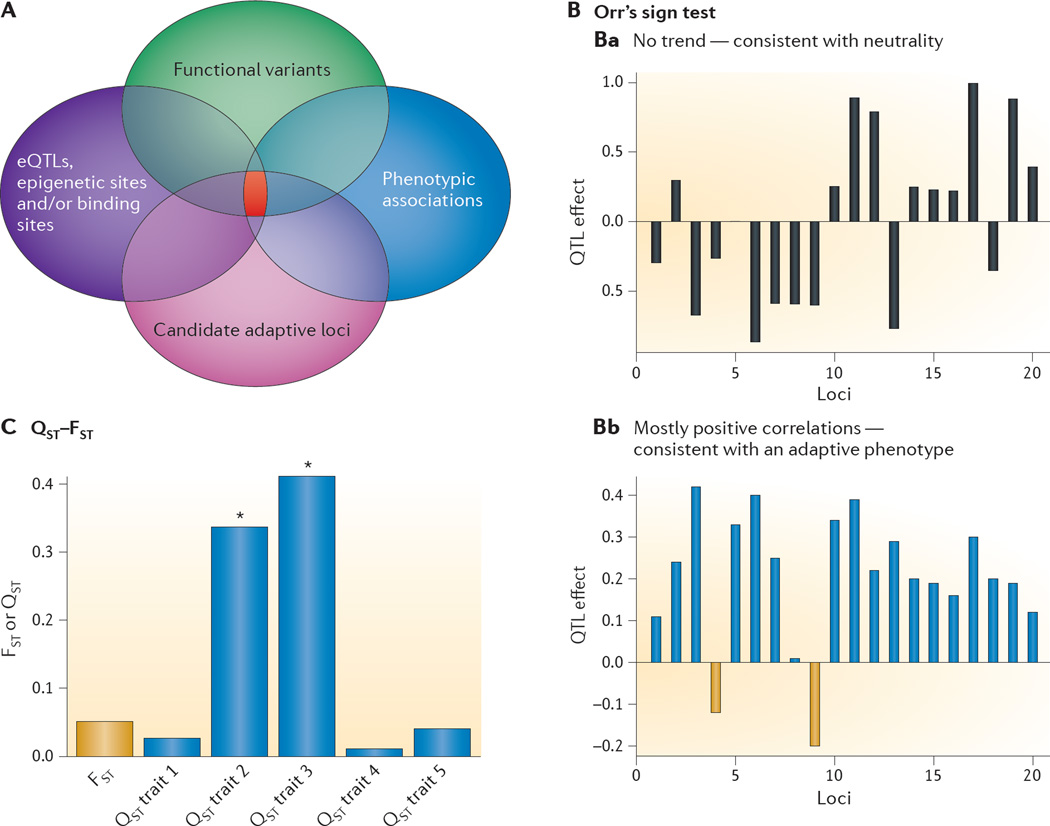
References
-
- Jobling MA, Hurles M, Tyler-Smith C. Human Evolutionary Genetics: Origins, Peoples and Disease. Garland Publishing; 2004.
-
-
Fraser HB. Gene expression drives local adaptation in humans. Genome Res. 2013;23:1089–1096. This study systematically evaluates the relative abundances of regulatory variation and coding variation in candidate adaptive regions and includes a novel method for identifying polygenic adaptation.
-
-
- Sabeti PC, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
