

Integration of new genes into cellular networks, and their structural maturation
- PMID: 24056411
- PMCID: PMC3832282
- DOI: 10.1534/genetics.113.152256
Integration of new genes into cellular networks, and their structural maturation
Abstract
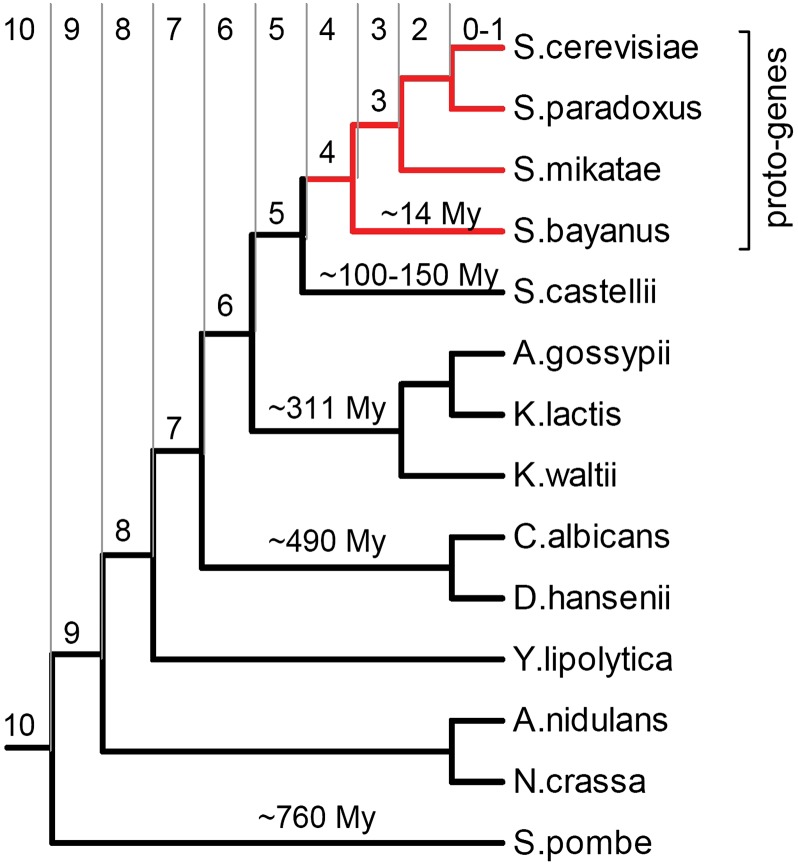
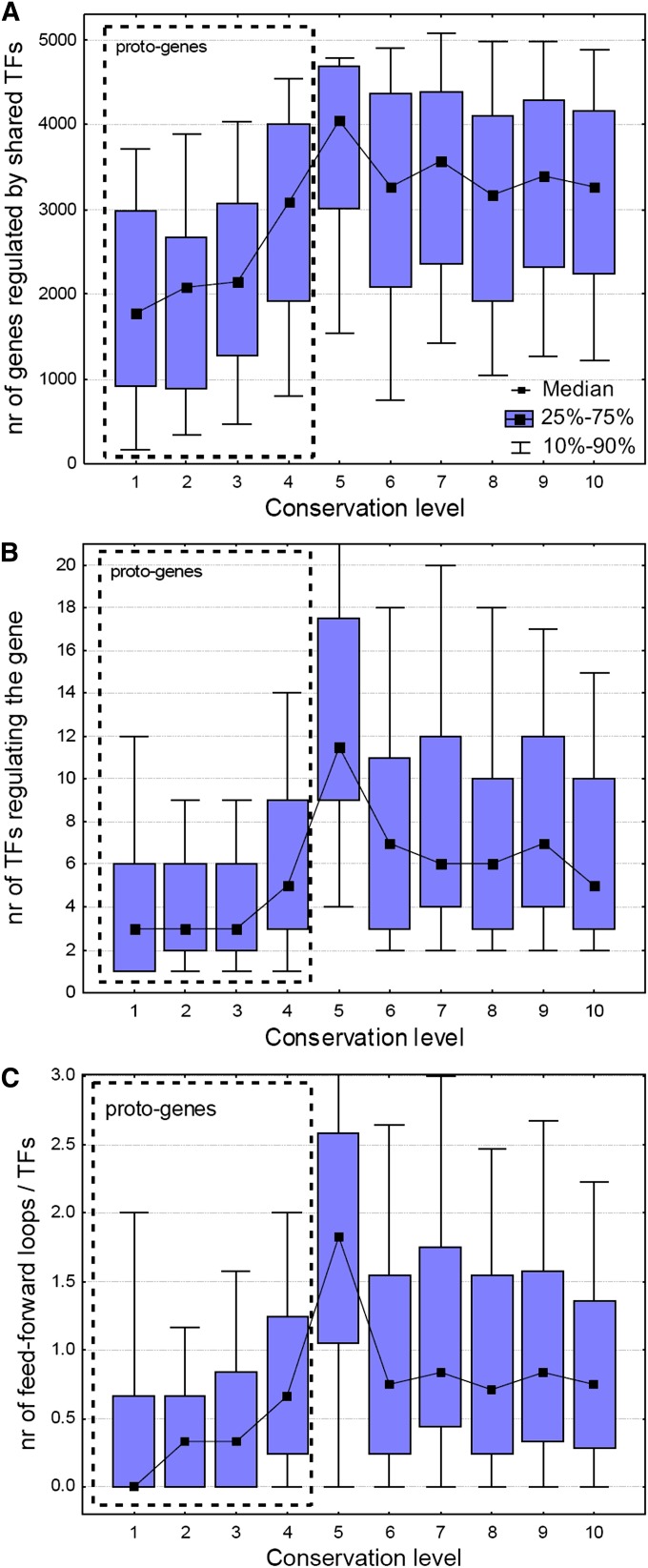
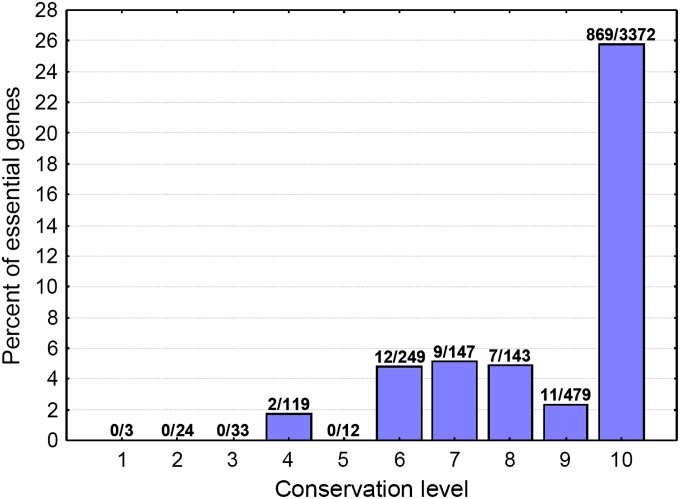
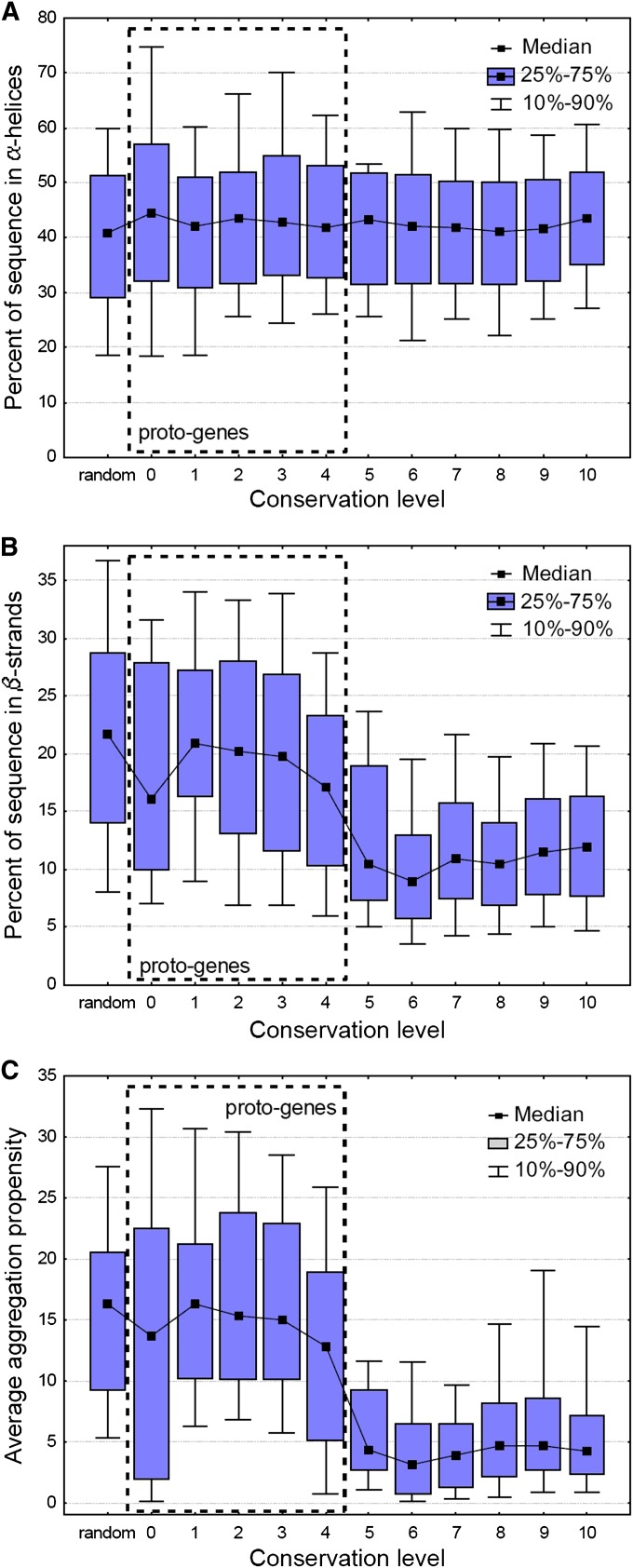
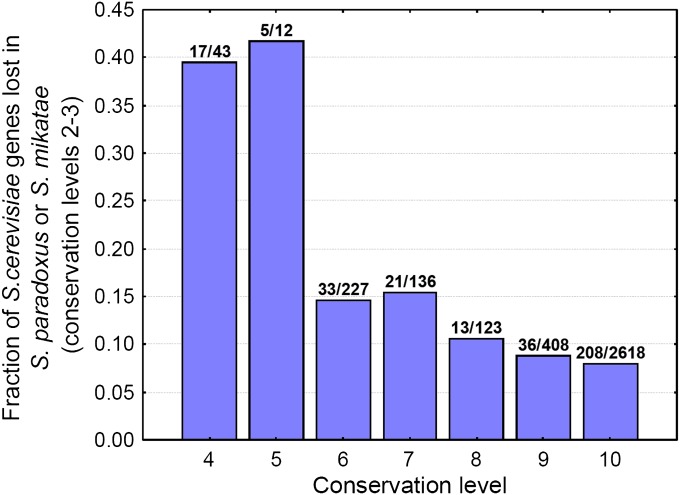
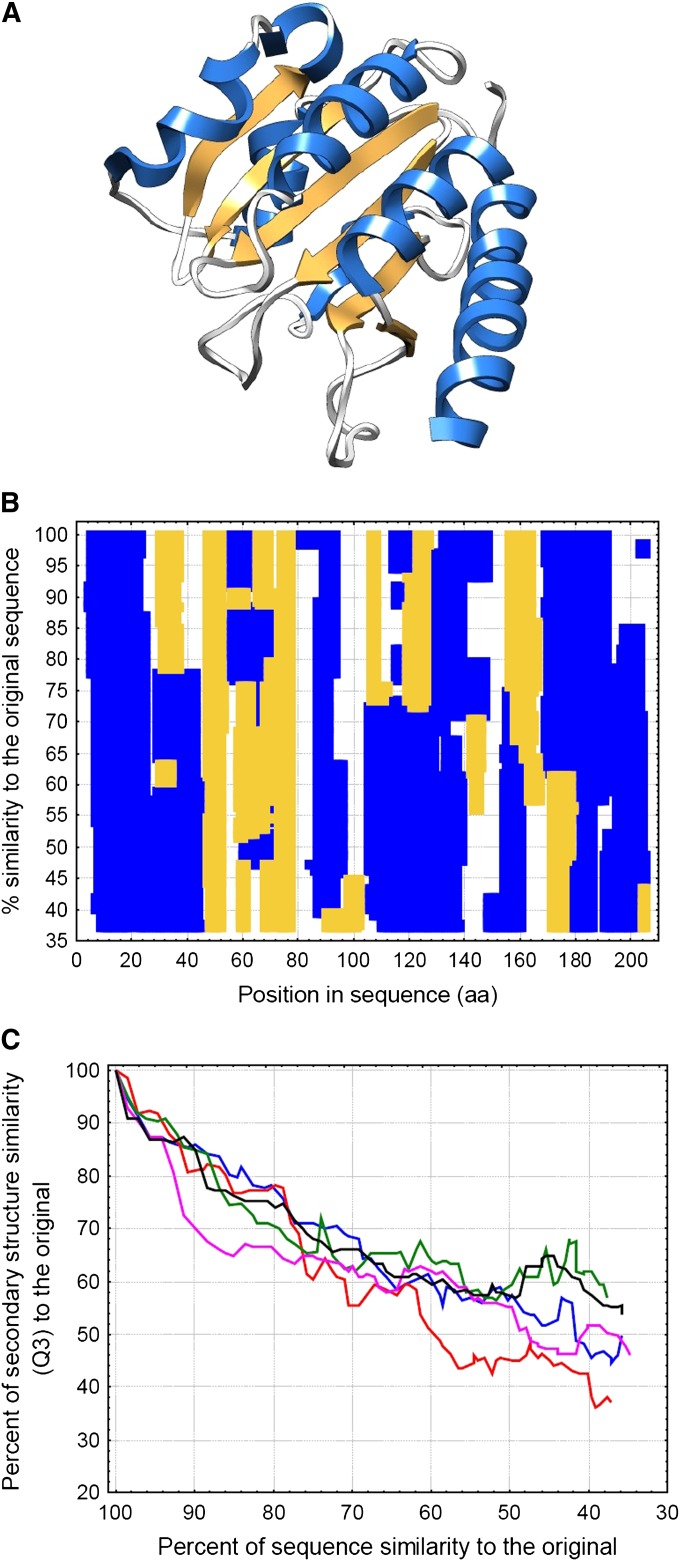
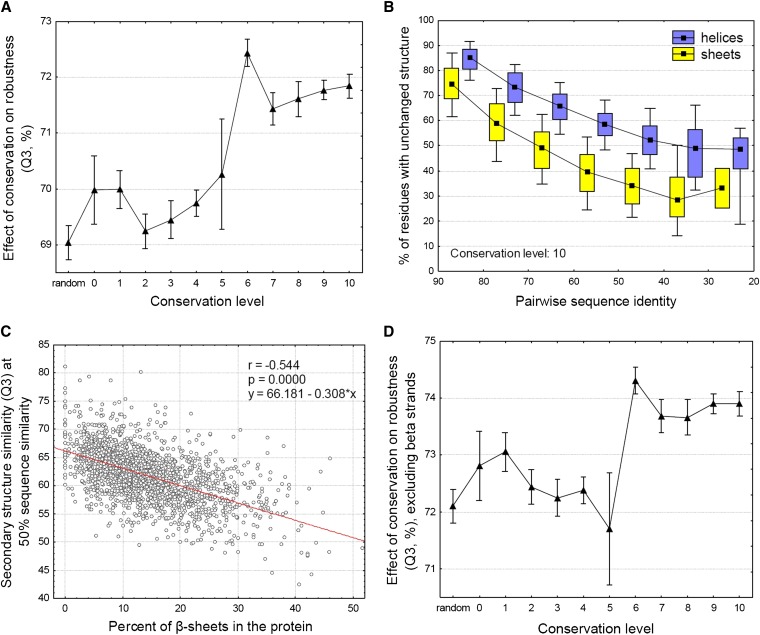
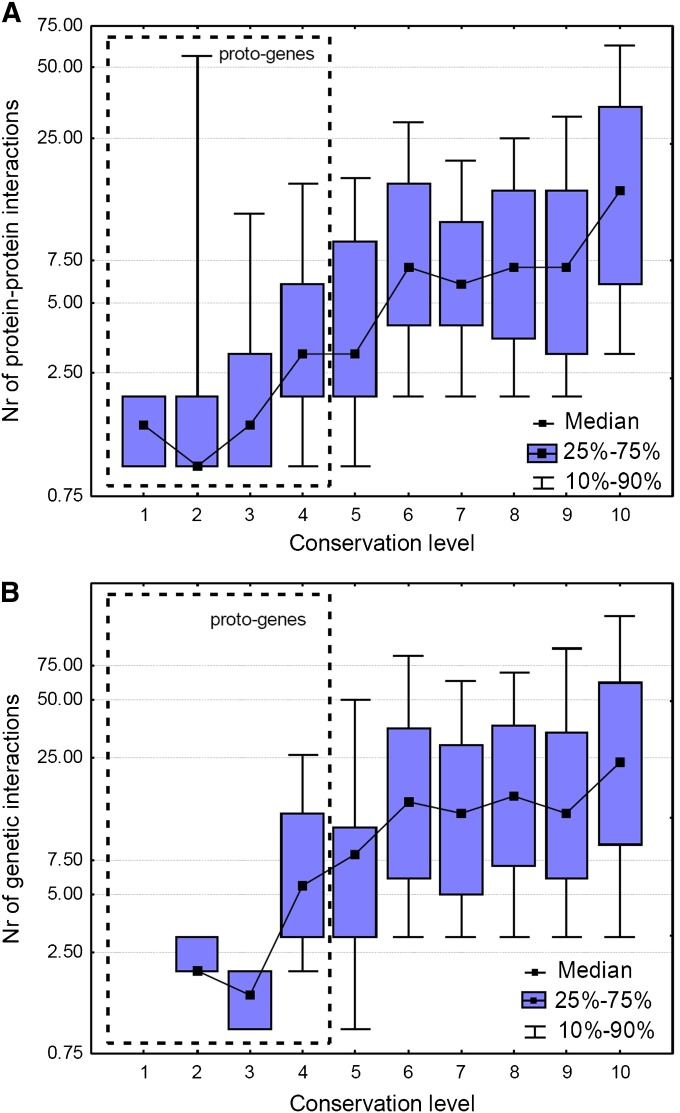
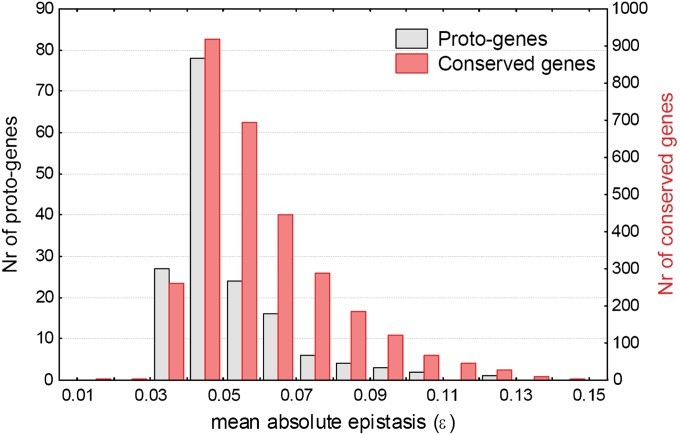
It has been recently discovered that new genes can originate de novo from noncoding DNA, and several biological traits including expression or sequence composition form a continuum from noncoding sequences to conserved genes. In this article, using yeast genes I test whether the integration of new genes into cellular networks and their structural maturation shows such a continuum by analyzing their changes with gene age. I show that 1) The number of regulatory, protein-protein, and genetic interactions increases continuously with gene age, although with very different rates. New regulatory interactions emerge rapidly within a few million years, while the number of protein-protein and genetic interactions increases slowly, with a rate of 2-2.25 × 10(-8)/year and 4.8 × 10(-8)/year, respectively. 2) Gene essentiality evolves relatively quickly: the youngest essential genes appear in proto-genes ∼14 MY old. 3) In contrast to interactions, the secondary structure of proteins and their robustness to mutations indicate that new genes face a bottleneck in their evolution: proto-genes are characterized by high β-strand content, high aggregation propensity, and low robustness against mutations, while conserved genes are characterized by lower strand content and higher stability, most likely due to the higher probability of gene loss among young genes and accumulation of neutral mutations.
Keywords: Saccharomyces cerevisiae; aggregation; de novo genes; protein–protein interaction; regulatory network; secondary structure.
Figures
References
-
- Bershtein S., Goldin K., Tawfik D. S., 2008. Intense neutral drifts yield robust and evolvable consensus proteins. J. Mol. Biol. 379: 1029–1044. - PubMed
-
- Bloom J. D., Drummond D. A., Arnold F. H., Wilke C. O., 2006a Structural determinants of the rate of protein evolution in yeast. Mol. Biol. Evol. 23: 1751–1761. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
