

Reversibility of neuropathology in Tay-Sachs-related diseases
- PMID: 24057669
- PMCID: PMC3888261
- DOI: 10.1093/hmg/ddt459
Reversibility of neuropathology in Tay-Sachs-related diseases
Abstract
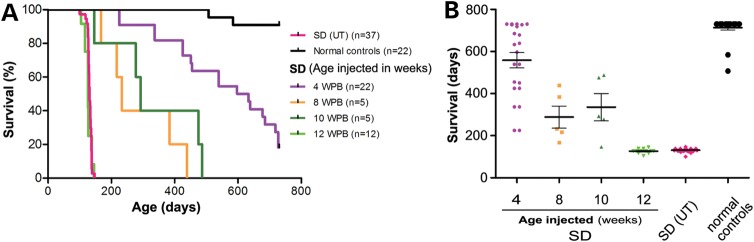
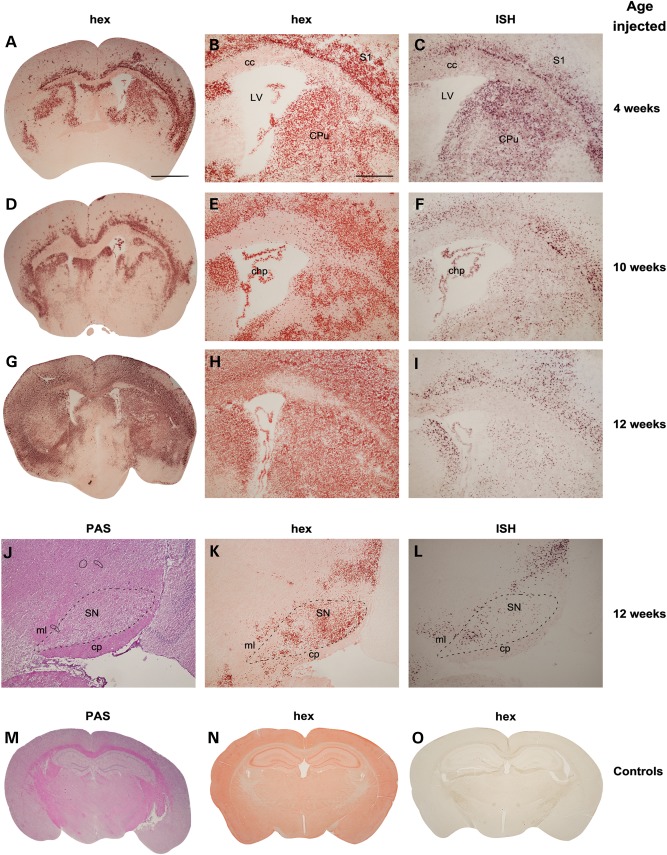
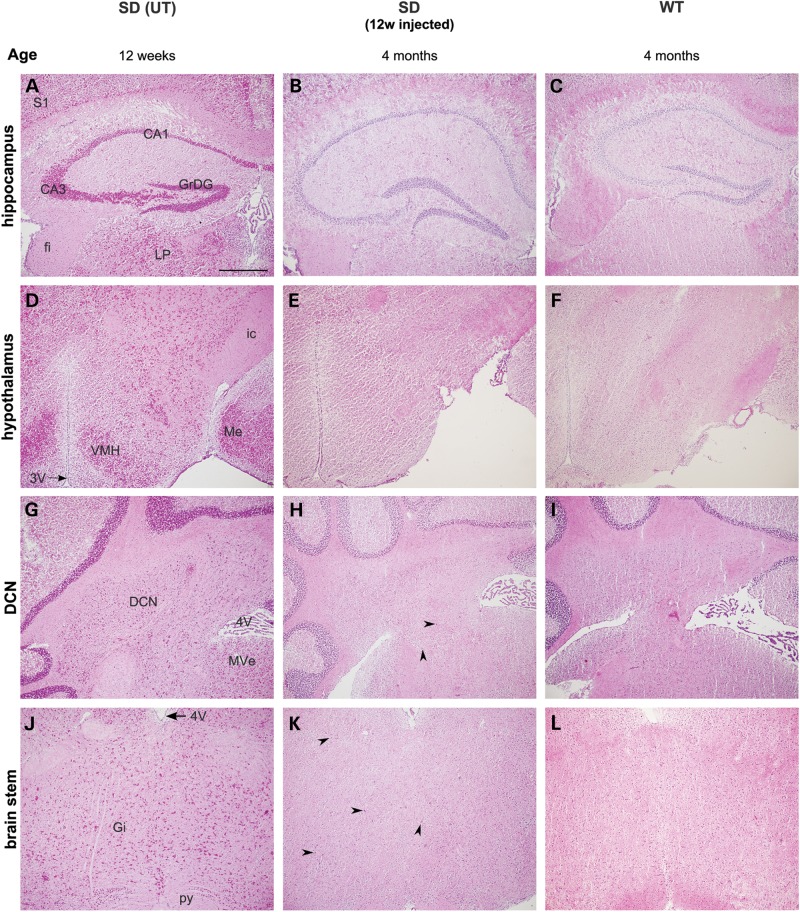
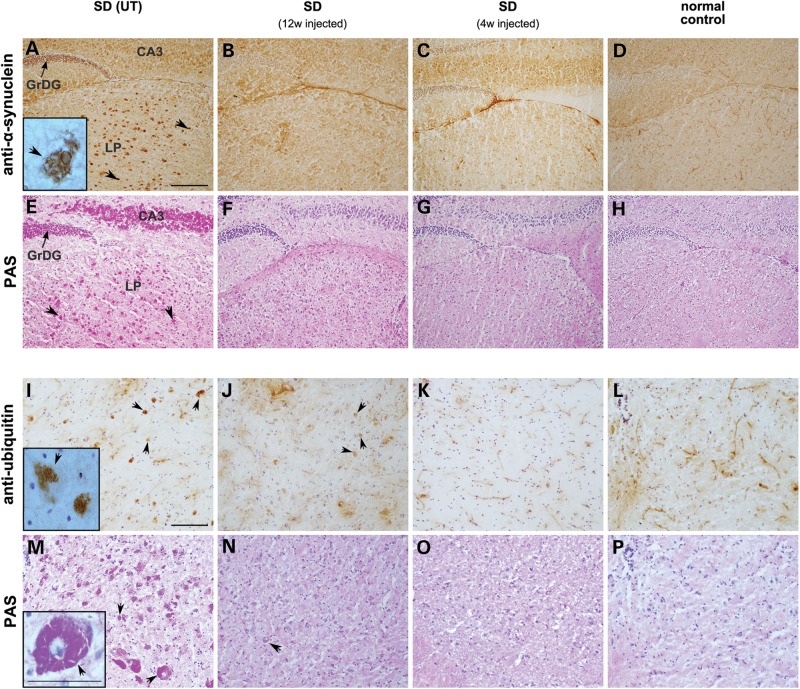
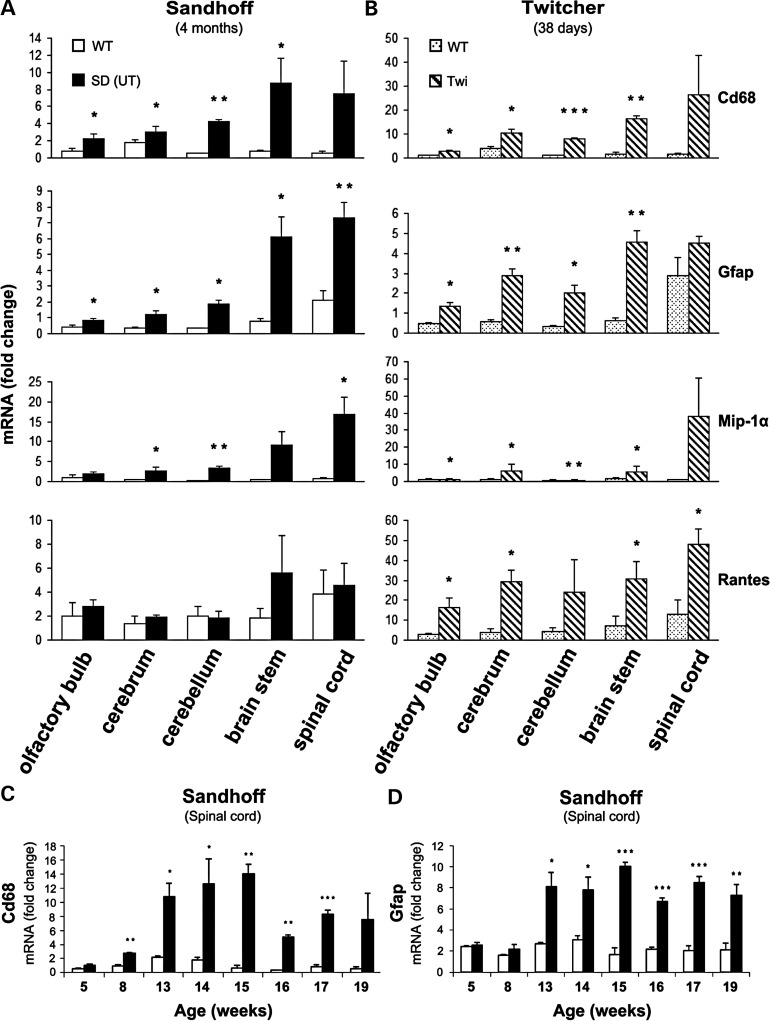
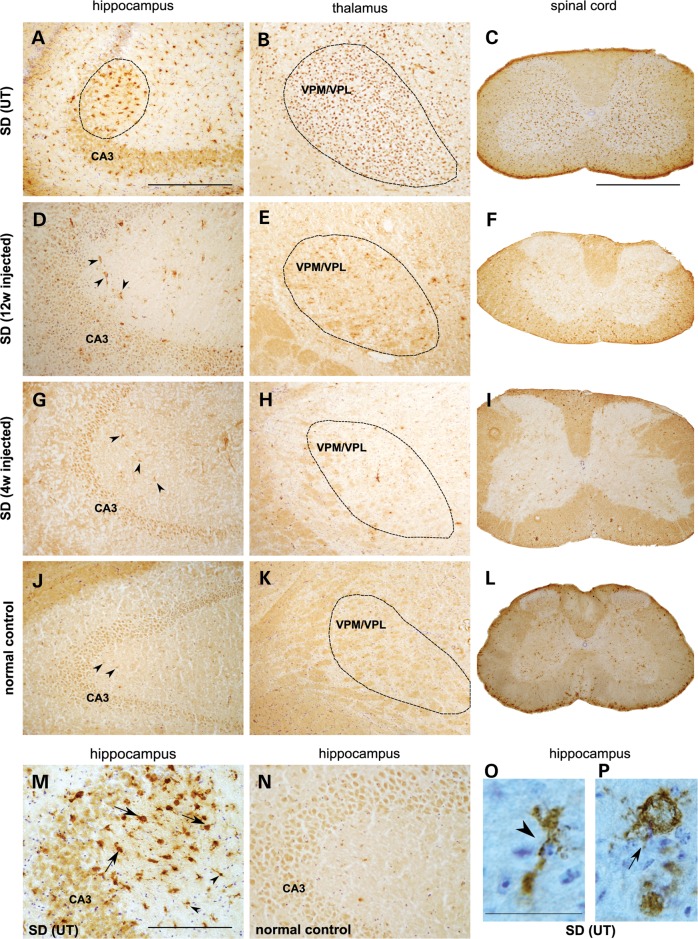
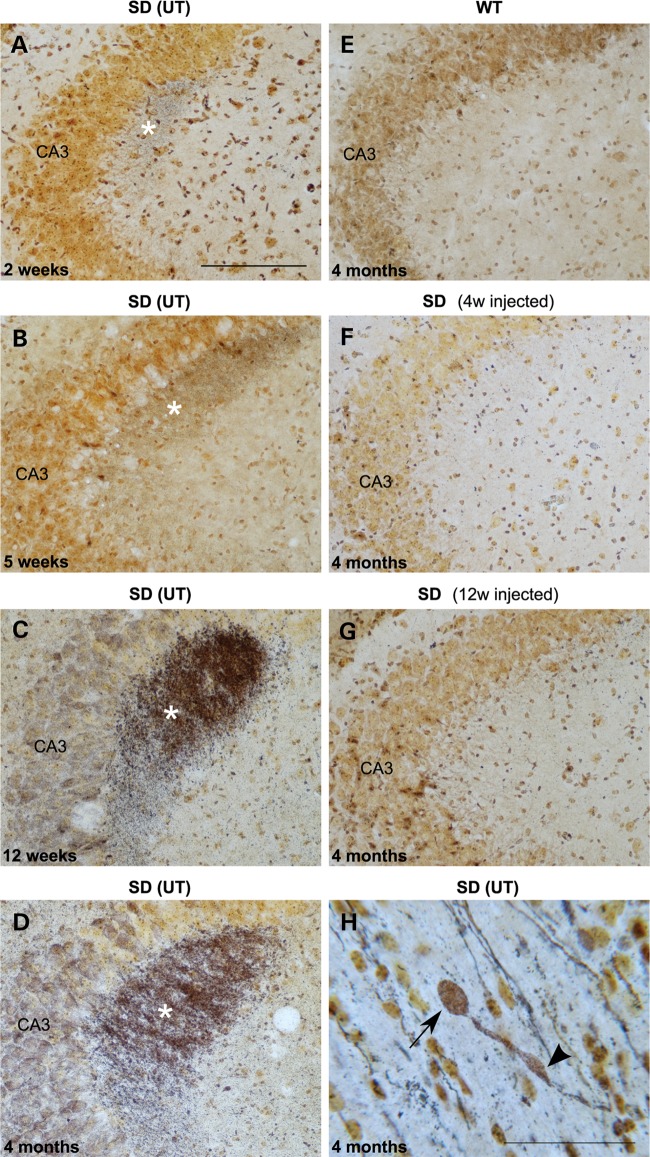
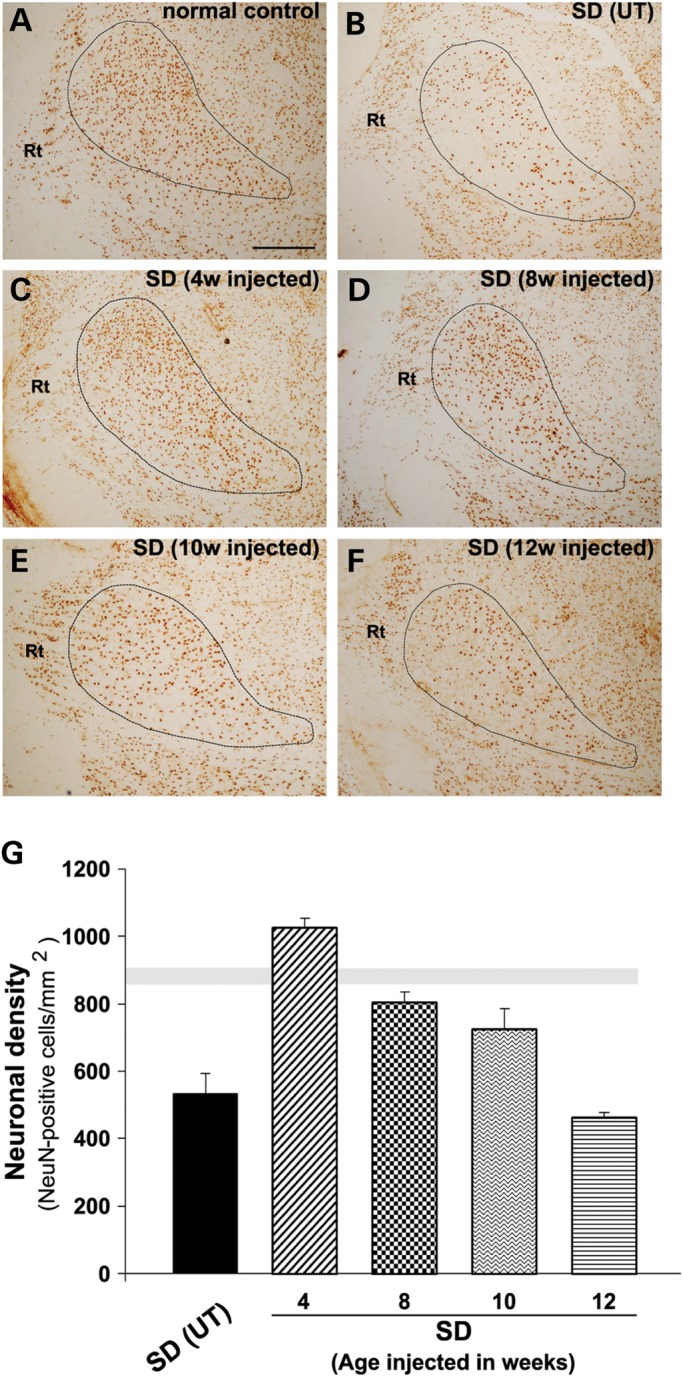
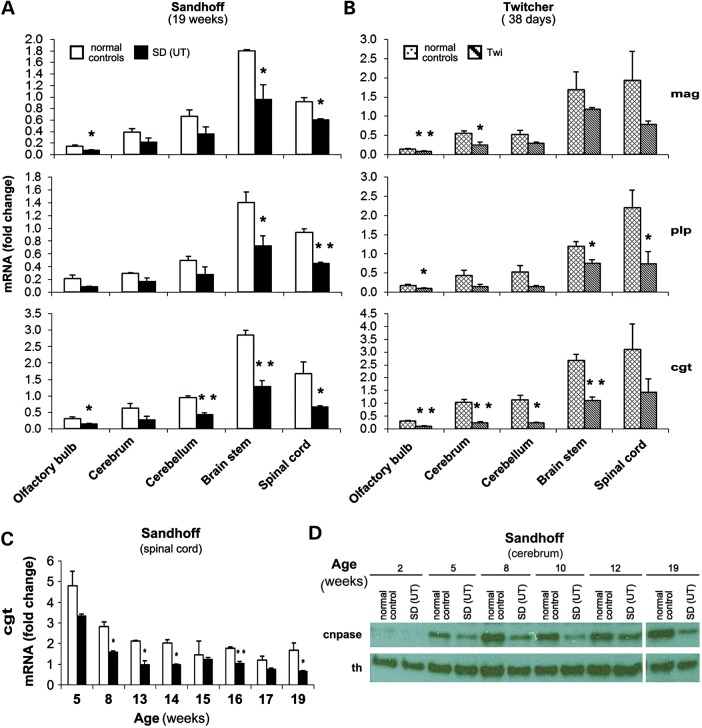
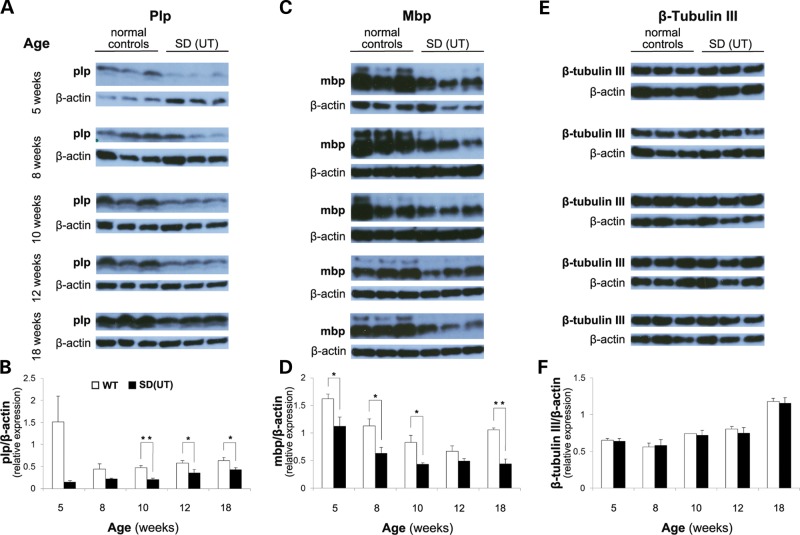
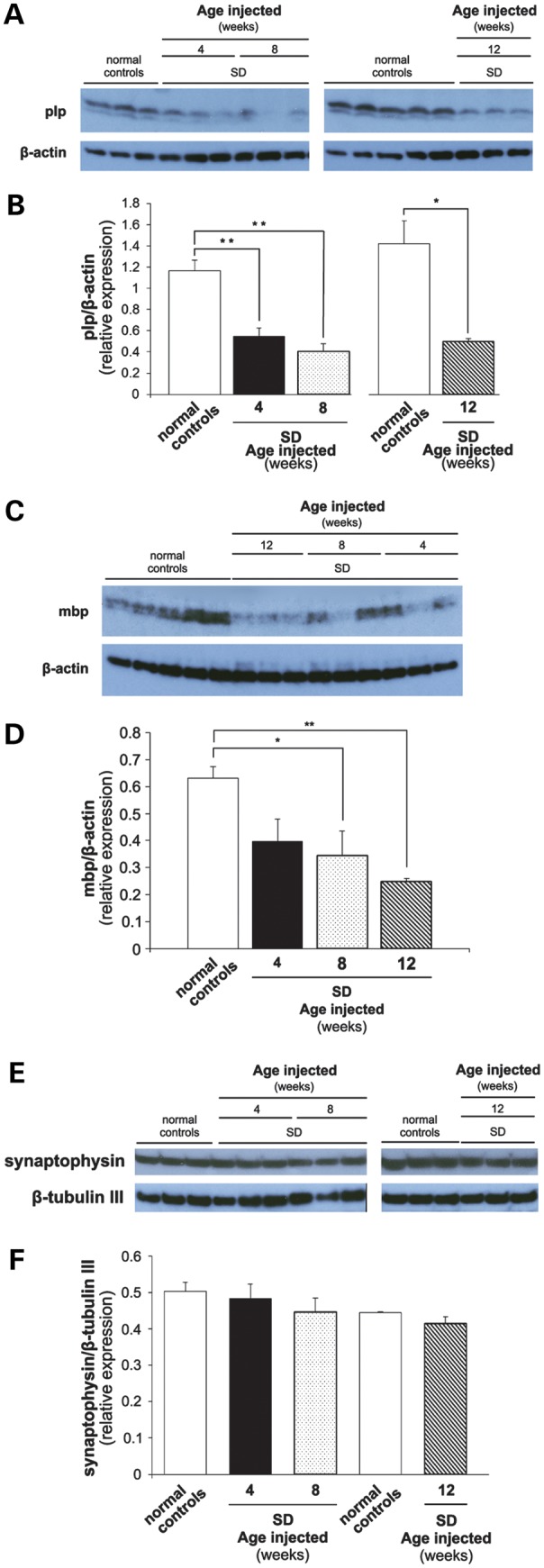
The GM2 gangliosidoses are progressive neurodegenerative disorders due to defects in the lysosomal β-N-acetylhexosaminidase system. Accumulation of β-hexosaminidases A and B substrates is presumed to cause this fatal condition. An authentic mouse model of Sandhoff disease (SD) with pathological characteristics resembling those noted in infantile GM2 gangliosidosis has been described. We have shown that expression of β-hexosaminidase by intracranial delivery of recombinant adeno-associated viral vectors to young adult SD mice can prevent many features of the disease and extends lifespan. To investigate the nature of the neurological injury in GM2 gangliosidosis and the extent of its reversibility, we have examined the evolution of disease in the SD mouse; we have moreover explored the effects of gene transfer delivered at key times during the course of the illness. Here we report greatly increased survival only when the therapeutic genes are expressed either before the disease is apparent or during its early manifestations. However, irrespective of when treatment was administered, widespread and abundant expression of β-hexosaminidase with consequent clearance of glycoconjugates, α-synuclein and ubiquitinated proteins, and abrogation of inflammatory responses and neuronal loss was observed. We also show that defects in myelination occur in early life and cannot be easily resolved when treatment is given to the adult brain. These results indicate that there is a limited temporal opportunity in which function and survival can be improved-but regardless of resolution of the cardinal pathological features of GM2 gangliosidosis, a point is reached when functional deterioration and death cannot be prevented.
Figures
References
-
- Sandhoff K., Andreae U., Jatzkewitz H. Deficient hexosaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs. Life Sci. 1968;7:283–288. doi:10.1016/0024-3205(68)90024-6. - DOI - PubMed
-
- Okada S., O'Brien J.S. Tay-Sachs disease: generalized absence of a beta-D-N-acetylhexosaminidase component. Science. 1969;165:698–700. doi:10.1126/science.165.3894.698. - DOI - PubMed
-
- Colzelmann E., Sandhoff K. AB variant of infantile GM2 gangliosidosis: deficiency of a factor necessary for stimulation of hexosaminidase A-catalyzed degradation of ganglioside GM2 and glycolipid GA2. Proc. Natl. Acad. Sci. USA. 1978;75:3979–3983. doi:10.1073/pnas.75.8.3979. - DOI - PMC - PubMed
-
- Kolodny E.H., Brady R.O., Volk B.W. Demonstration of an alteration of ganglioside metabolism in Tay-Sachs disease. Biochem. Biophys. Res. Commun. 1969;37:526–531. doi:10.1016/0006-291X(69)90947-4. - DOI - PubMed
-
- Sandhoff K. Variation of beta-N-acetylhexosaminidase-pattern in Tay-Sachs disease. FEBS Lett. 1969;4:351–354. doi:10.1016/0014-5793(69)80274-7. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
