

Mechanistic insight into the relationship between N-terminal acetylation of α-synuclein and fibril formation rates by NMR and fluorescence
- PMID: 24058647
- PMCID: PMC3776725
- DOI: 10.1371/journal.pone.0075018
Mechanistic insight into the relationship between N-terminal acetylation of α-synuclein and fibril formation rates by NMR and fluorescence
Abstract
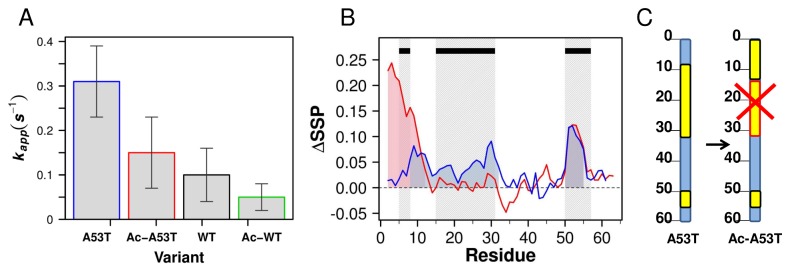
Aggregation of α-synuclein (αSyn), the primary protein component in Lewy body inclusions of patients with Parkinson's disease, arises when the normally soluble intrinsically disordered protein converts to amyloid fibrils. In this work, we provide a mechanistic view of the role of N-terminal acetylation on fibrillation by first establishing a quantitative relationship between monomer secondary structural propensity and fibril assembly kinetics, and secondly by demonstrating in the N-terminal acetylated form of the early onset A53T mutation, that N-terminal transient helices formed and/or inhibited by N-terminal acetylation modulate the fibril assembly rates. Using NMR chemical shifts and fluorescence experiments, we report that secondary structural propensity in residues 5-8, 14-31, and 50-57 are highly correlated to fibril growth rate. A four-way comparison of secondary structure propensity and fibril growth rates of N-terminally acetylated A53T and WT αSyn with non-acetylated A53T and WT αSyn present novel mechanistic insight into the role of N-terminal acetylation in amyloid fibril formation. We show that N-terminal acetylation inhibits the formation of the "fibrillation promoting" transient helix at residues 14-31 resulting from the A53T mutation in the non-acetylated variant and supports the formation of the "fibrillation inhibiting" transient helix in residues 1-12 thereby resulting in slower fibrillation rates relative to the previously studied non-acetylated A53T variant. Our results highlight the critical interplay of the region-specific transient secondary structure of the N-terminal region with fibrillation, and the inhibitory role of the N-terminal acetyl group in fibril formation.
Conflict of interest statement
Figures



References
-
- Lansbury PT Jr. (1999) Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci U S A 96: 3342-3344. doi:10.1073/pnas.96.7.3342. PubMed: 10097040. - DOI - PMC - PubMed
-
- Perutz MF (1999) Glutamine repeats and neurodegenerative diseases: molecular aspects. Trends Biochem Sci 24: 58-63. doi:10.1016/S0968-0004(98)01350-4. PubMed: 10098399. - DOI - PubMed
-
- Dobson CM (1999) Protein misfolding, evolution and disease. Trends Biochem Sci 24: 329-332. doi:10.1016/S0968-0004(99)01445-0. PubMed: 10470028. - DOI - PubMed
-
- Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75: 333-366. doi:10.1146/annurev.biochem.75.101304.123901. PubMed: 16756495. - DOI - PubMed
-
- Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10 Suppl: S10-S17. doi:10.1038/nm1066. PubMed: 15272267. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
