

DNA damage tolerance: a double-edged sword guarding the genome
- PMID: 24058901
- PMCID: PMC3779140
- DOI: 10.3978/j.issn.2218-676X.2013.04.01
DNA damage tolerance: a double-edged sword guarding the genome
Abstract
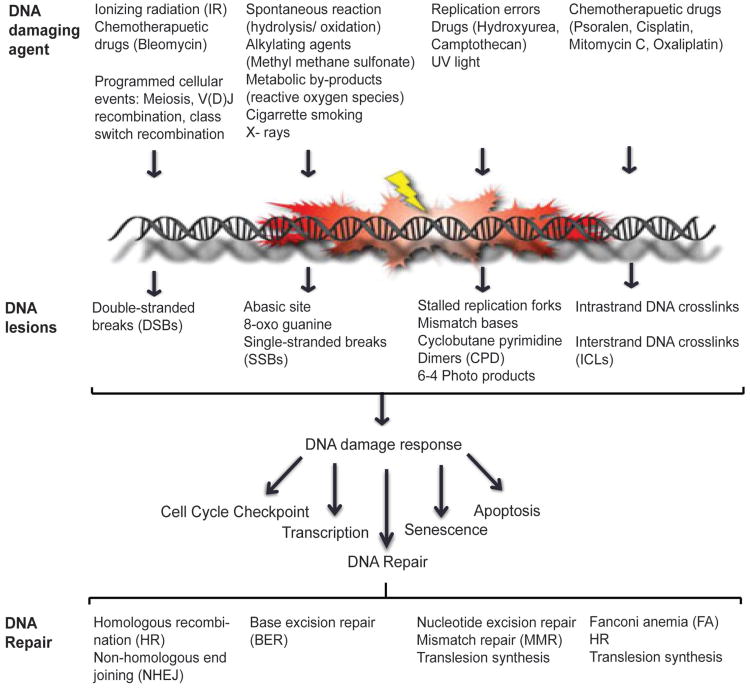
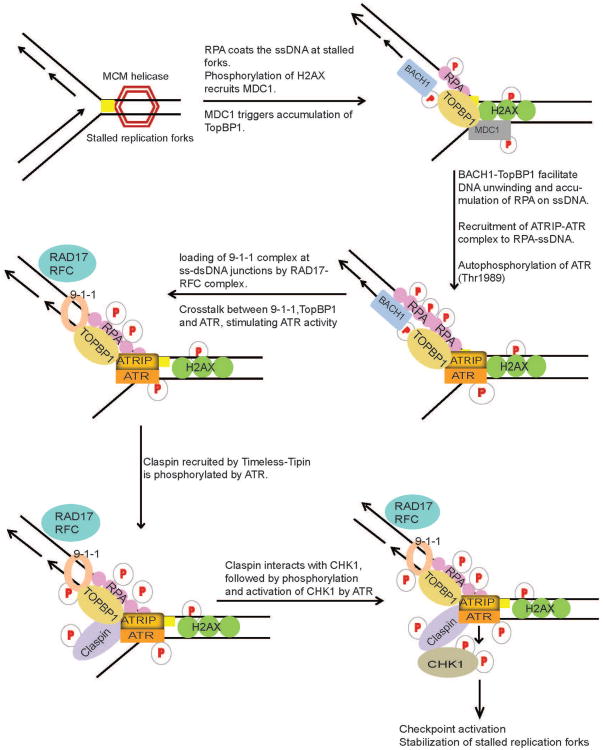
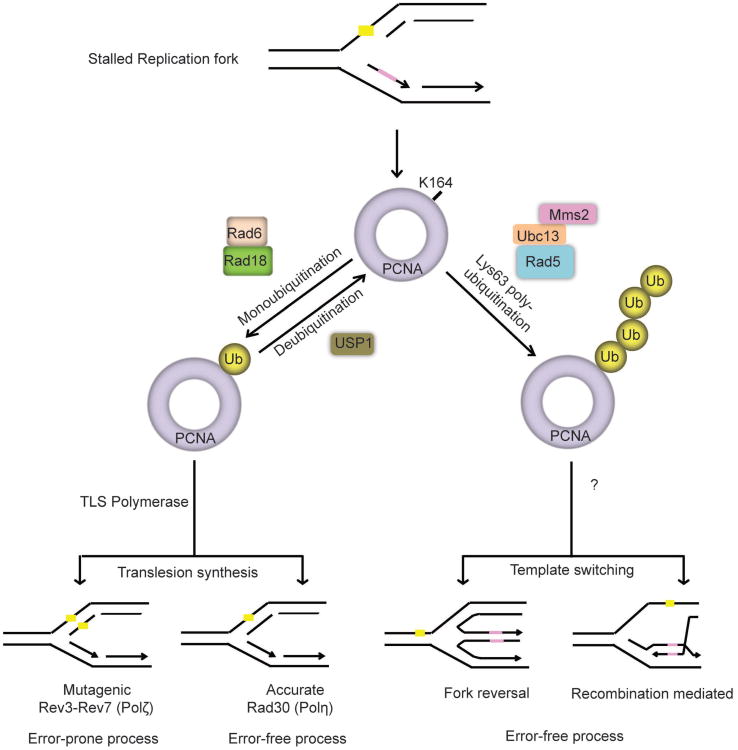
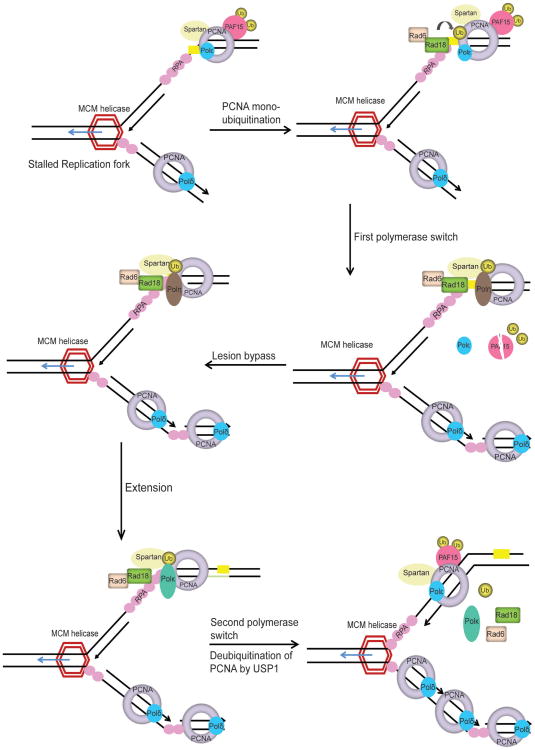
Preservation of genome integrity is an essential process for cell homeostasis. During the course of life of a single cell, the genome is constantly damaged by endogenous and exogenous agents. To ensure genome stability, cells use a global signaling network, namely the DNA damage response (DDR) to sense and repair DNA damage. DDR senses different types of DNA damage and coordinates a response that includes activation of transcription, cell cycle control, DNA repair pathways, apoptosis, senescence, and cell death. Despite several repair mechanisms that repair different types of DNA lesions, it is likely that the replication machinery would still encounter lesions that are mis-repaired or not repaired. Replication of damaged genome would result in high frequency of fork collapse and genome instability. In this scenario, the cells employ the DNA damage tolerance (DDT) pathway that recruits a specialized low fidelity translesion synthesis (TLS) polymerase to bypass the lesions for repair at a later time point. Thus, DDT is not a repair pathway per se, but provides a mechanism to tolerate DNA lesions during replication thereby increasing survival and preventing genome instability. Paradoxically, DDT process is also associated with increased mutagenesis, which can in turn drive the cell to cancer development. Thus, DDT process functions as a double-edged sword guarding the genome. In this review, we will discuss the replication stress induced DNA damage-signaling cascade, the stabilization and rescue of stalled replication forks by the DDT pathway and the effect of the DDT pathway on cancer.
Keywords: DNA damage tolerance (DDT); proliferating cell nuclear antigen (PCNA); replicative DNA polymerase; stalled replication forks; translesion polymerase; translesion synthesis (TLS).
Conflict of interest statement
Figures
References
-
- Kasparek TR, Humphrey TC. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin Cell Dev Biol. 2011;22:886–97. - PubMed
-
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–9. - PubMed
-
- Moraes MC, Neto JB, Menck CF. DNA repair mechanisms protect our genome from carcinogenesis. Front Biosci. 2012;17:1362–88. - PubMed
-
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–85. - PubMed
-
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous