

DNA methylation and differentiation: silencing, upregulation and modulation of gene expression
- PMID: 24059801
- PMCID: PMC3864898
- DOI: 10.2217/epi.13.43
DNA methylation and differentiation: silencing, upregulation and modulation of gene expression
Abstract
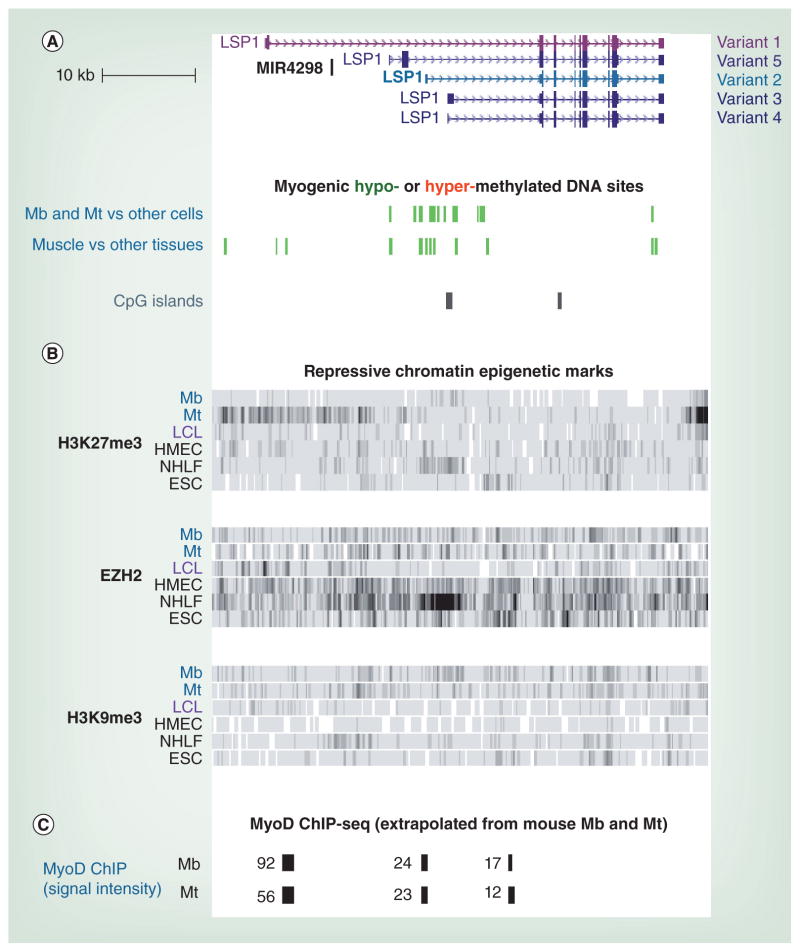
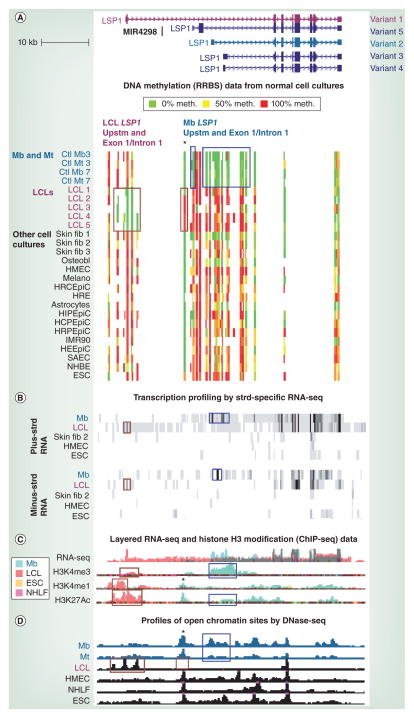
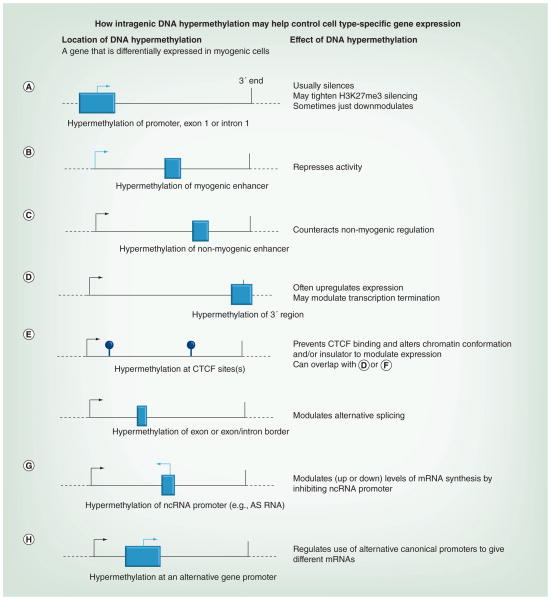
Differentiation-related DNA methylation is receiving increasing attention, partly owing to new, whole-genome analyses. These revealed that cell type-specific differential methylation in gene bodies is more frequent than in promoters. We review new insights into the functionality of DNA methylation during differentiation, with emphasis on the methylomes of myoblasts, myotubes and skeletal muscle versus non-muscle samples. Biostatistical analyses of data from reduced representation bisulfite sequencing are discussed. Lastly, a model is presented for how promoter and intragenic DNA hypermethylation affect gene expression, including increasing the efficiency of polycomb silencing at some promoters, downmodulating other promoters rather than silencing them, counteracting enhancers with heterologous specificity, altering chromatin conformation by inhibiting the binding of CTCF, modulating mRNA transcript levels by inhibiting overlapping promoters of noncoding RNA genes or by regulating the use of alternative mRNA promoters, modulating transcription termination, regulating alternative splicing and acting as barriers to the spread of activating chromatin.
Figures
References
-
- Ndlovu MN, Denis H, Fuks F. Exposing the DNA methylome iceberg. Trends Biochem Sci. 2011;36(7):381–387. - PubMed
Website
-
- UCSC Genome Bioinformatics. http://genome.ucsc.edu.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources