

STAT3 regulation by S-nitrosylation: implication for inflammatory disease
- PMID: 24063605
- PMCID: PMC4026100
- DOI: 10.1089/ars.2013.5223
STAT3 regulation by S-nitrosylation: implication for inflammatory disease
Abstract
Aims: S-nitrosylation and S-glutathionylation, redox-based modifications of protein thiols, are recently emerging as important signaling mechanisms. In this study, we assessed S-nitrosylation-based regulation of Janus-activated kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) pathway that plays critical roles in immune/inflammatory responses and tumorigenesis.
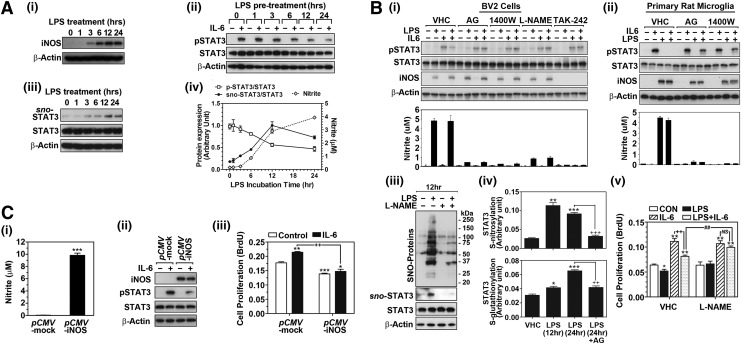
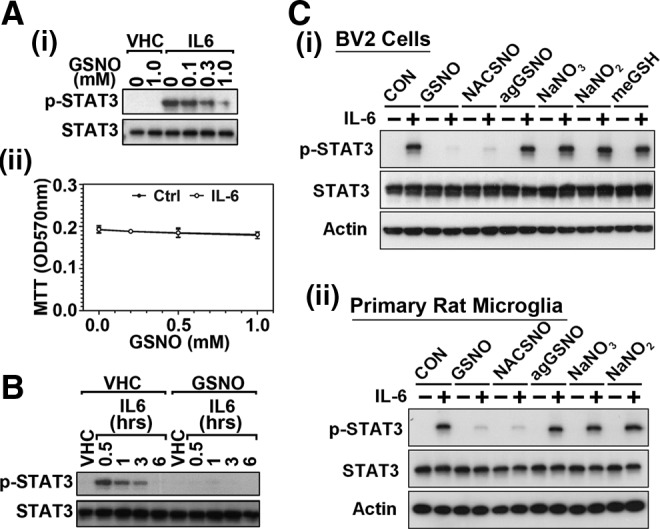
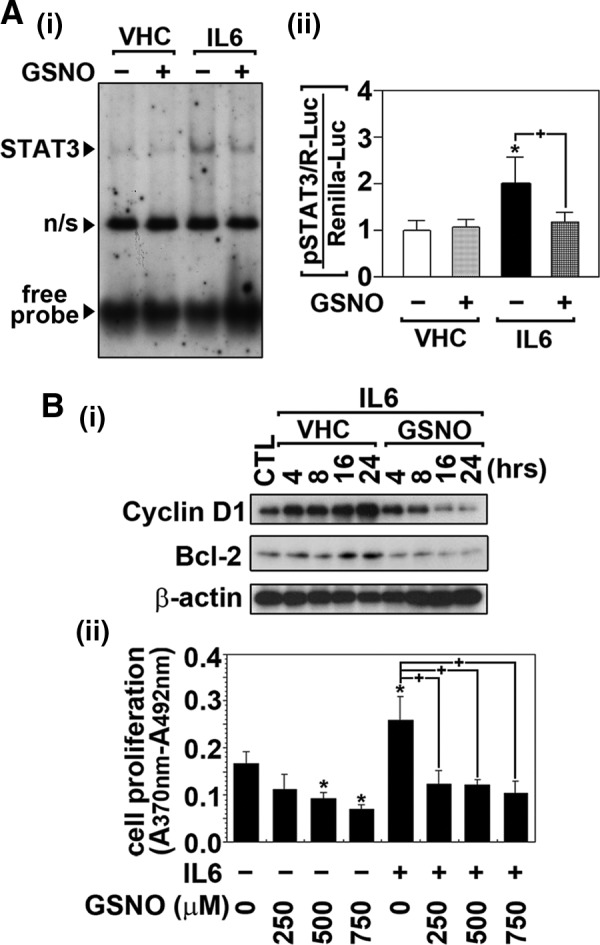
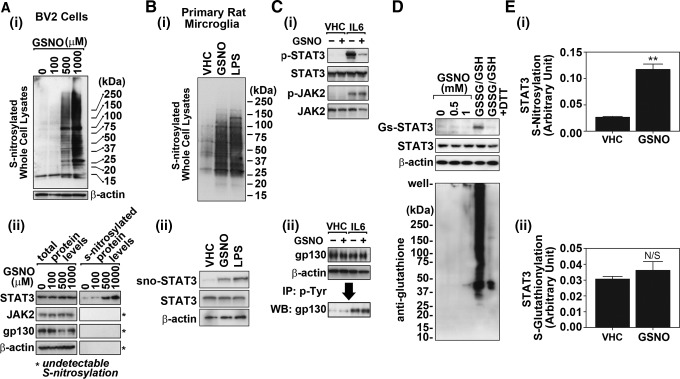
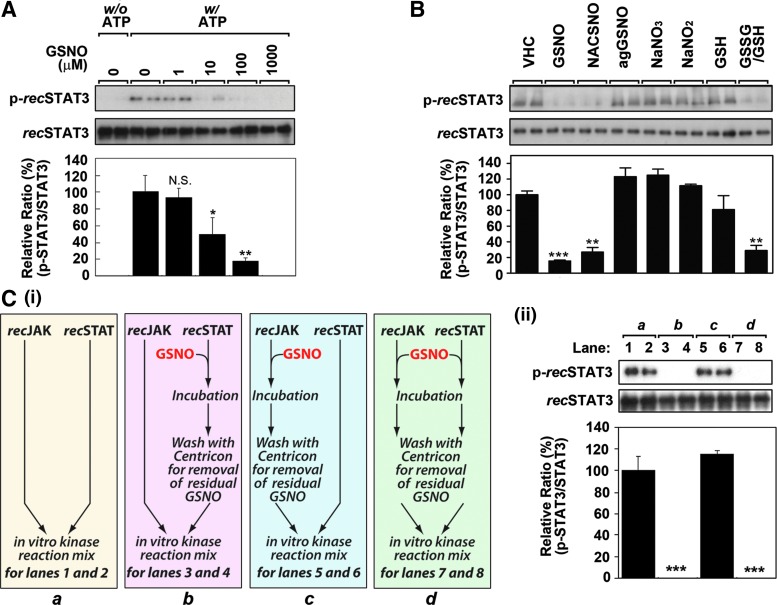
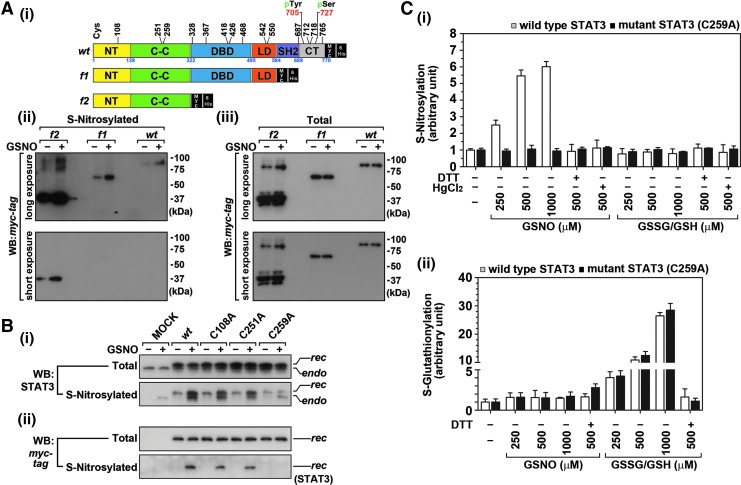
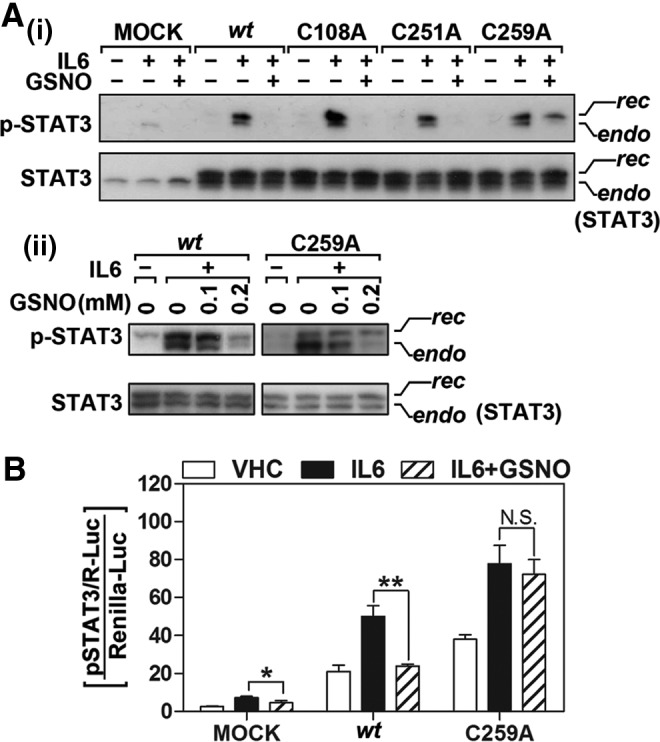
Results: Our studies show that STAT3 in stimulated microglia underwent two distinct redox-dependent modifications, S-nitrosylation and S-glutathionylation. STAT3 S-nitrosylation was associated with inducible nitric oxide synthase (iNOS)-produced nitric oxide (NO) and S-nitrosoglutathione (GSNO), whereas S-glutathionylation of STAT3 was associated with cellular oxidative stress. NO produced by iNOS or treatment of microglia with exogenous GSNO inhibited STAT3 activation via inhibiting STAT3 phosphorylation (Tyr(705)). Consequently, the interleukin-6 (IL-6)-induced microglial proliferation and associated gene expressions were also reduced. In cell-free kinase assay using purified JAK2 and STAT3, STAT3 phosphorylation was inhibited by its selective preincubation with GSNO, but not by preincubation of JAK2 with GSNO, indicating that GSNO-mediated mechanisms inhibit STAT3 phosphorylation through S-nitrosylation of STAT3 rather than JAK2. In this study, we identified that Cys(259) was the target Cys residue of GSNO-mediated S-nitrosylation of STAT3. The replacement of Cys(259) residue with Ala abolished the inhibitory role of GSNO in IL-6-induced STAT3 phosphorylation and transactivation, suggesting the role of Cys(259) S-nitrosylation in STAT3 phosphorylation.
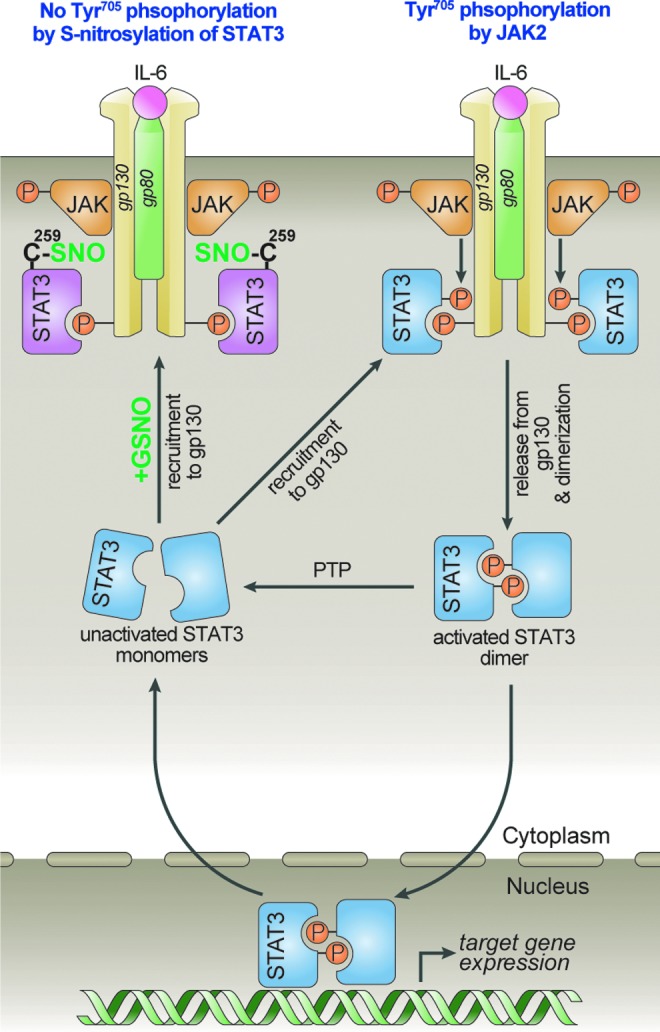
Innovation: Microglial proliferation is regulated by NO via S-nitrosylation of STAT3 (Cys(259)) and inhibition of STAT3 (Tyr(705)) phosphorylation.
Conclusion: Our results indicate the regulation of STAT3 by NO-based post-translational modification (S-nitrosylation). These findings have important implications for the development of new therapeutics targeting STAT3 for treating diseases associated with inflammatory/immune responses and abnormal cell proliferation, including cancer.
Figures
Similar articles
-
S-Nitrosoglutathione-mediated STAT3 regulation in efficacy of radiotherapy and cisplatin therapy in head and neck squamous cell carcinoma.Redox Biol. 2015 Dec;6:41-50. doi: 10.1016/j.redox.2015.07.001. Epub 2015 Jul 2. Redox Biol. 2015. PMID: 26177470 Free PMC article.
-
STAT3 Regulation By S-Nitrosylation: Implication In Cancer.Redox Biol. 2015 Aug;5:416-417. doi: 10.1016/j.redox.2015.09.021. Epub 2015 Dec 30. Redox Biol. 2015. PMID: 28162277
-
The role of the JAK2-STAT3 pathway in pro-inflammatory responses of EMF-stimulated N9 microglial cells.J Neuroinflammation. 2010 Sep 9;7:54. doi: 10.1186/1742-2094-7-54. J Neuroinflammation. 2010. PMID: 20828402 Free PMC article.
-
The redox pathway of S-nitrosoglutathione, glutathione and nitric oxide in cell to neuron communications.Free Radic Res. 1999 Dec;31(6):641-50. doi: 10.1080/10715769900301211. Free Radic Res. 1999. PMID: 10630687 Review.
-
Glutathione in Protein Redox Modulation through S-Glutathionylation and S-Nitrosylation.Molecules. 2021 Jan 15;26(2):435. doi: 10.3390/molecules26020435. Molecules. 2021. PMID: 33467703 Free PMC article. Review.
Cited by
-
Targeting Cervical Cancer Stem Cells by Phytochemicals.Curr Med Chem. 2024;31(32):5222-5254. doi: 10.2174/0109298673281823231222065616. Curr Med Chem. 2024. PMID: 38288813 Review.
-
STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment.Int J Mol Sci. 2018 Jun 16;19(6):1787. doi: 10.3390/ijms19061787. Int J Mol Sci. 2018. PMID: 29914167 Free PMC article. Review.
-
GSNO promotes functional recovery in experimental TBI by stabilizing HIF-1α.Behav Brain Res. 2018 Mar 15;340:63-70. doi: 10.1016/j.bbr.2016.10.037. Epub 2016 Oct 22. Behav Brain Res. 2018. PMID: 27780722 Free PMC article.
-
S-Nitrosoglutathione-mediated STAT3 regulation in efficacy of radiotherapy and cisplatin therapy in head and neck squamous cell carcinoma.Redox Biol. 2015 Dec;6:41-50. doi: 10.1016/j.redox.2015.07.001. Epub 2015 Jul 2. Redox Biol. 2015. PMID: 26177470 Free PMC article.
-
Hepatocellular Protein Arginine Methyltransferase 1 Suppresses Alcohol-Induced Hepatocellular Carcinoma Formation by Inhibition of Inducible Nitric Oxide Synthase.Hepatol Commun. 2020 Mar 4;4(6):790-808. doi: 10.1002/hep4.1488. eCollection 2020 Jun. Hepatol Commun. 2020. PMID: 32490317 Free PMC article.
References
-
- Baker TL, Booden MA, and Buss JE. S-Nitrosocysteine increases palmitate turnover on Ha-Ras in NIH 3T3 cells. J Biol Chem 275: 22037–22047, 2000 - PubMed
-
- Block ML. and Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76: 77–98, 2005 - PubMed
-
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, and Darnell JE., Jr.Stat3 as an oncogene. Cell 98: 295–303, 1999 - PubMed
-
- Campbell IL. Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res Brain Res Rev 48: 166–177, 2005 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
