

Differential involvement of E2A-corepressor interactions in distinct leukemogenic pathways
- PMID: 24064250
- PMCID: PMC3874172
- DOI: 10.1093/nar/gkt855
Differential involvement of E2A-corepressor interactions in distinct leukemogenic pathways
Abstract
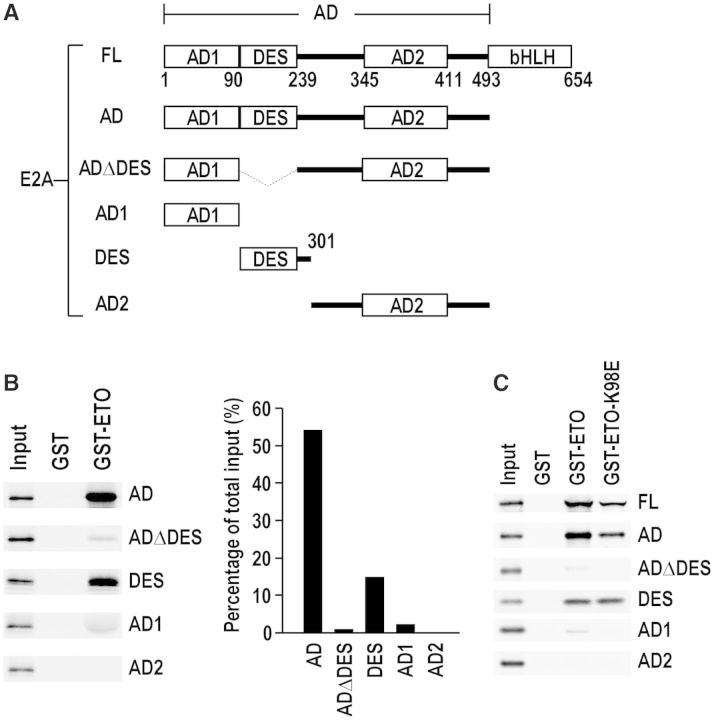
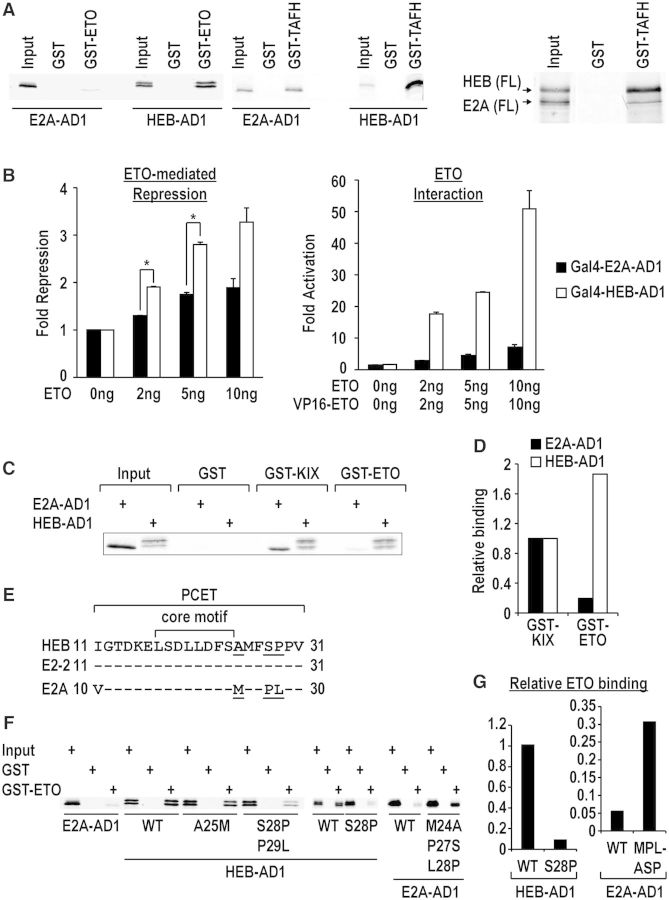
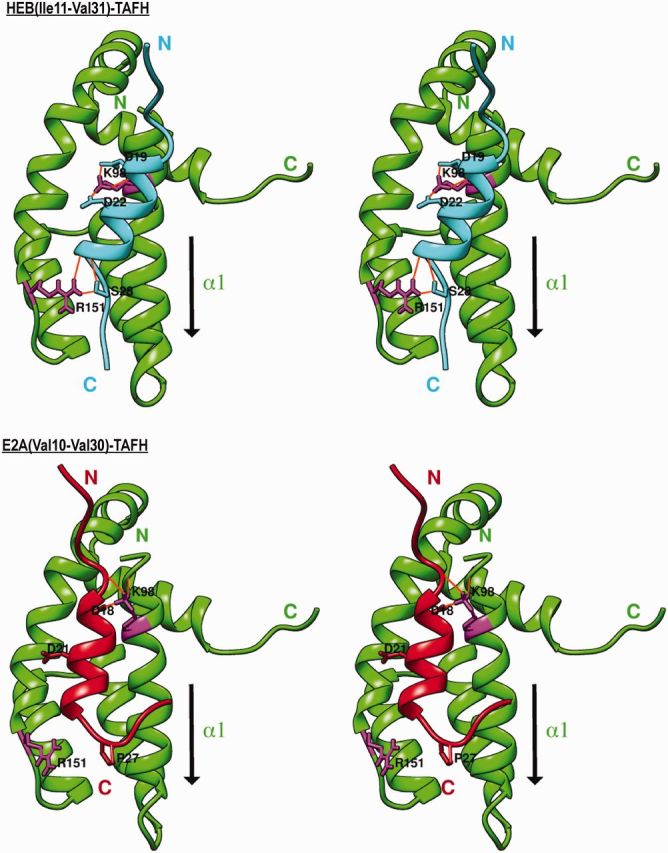
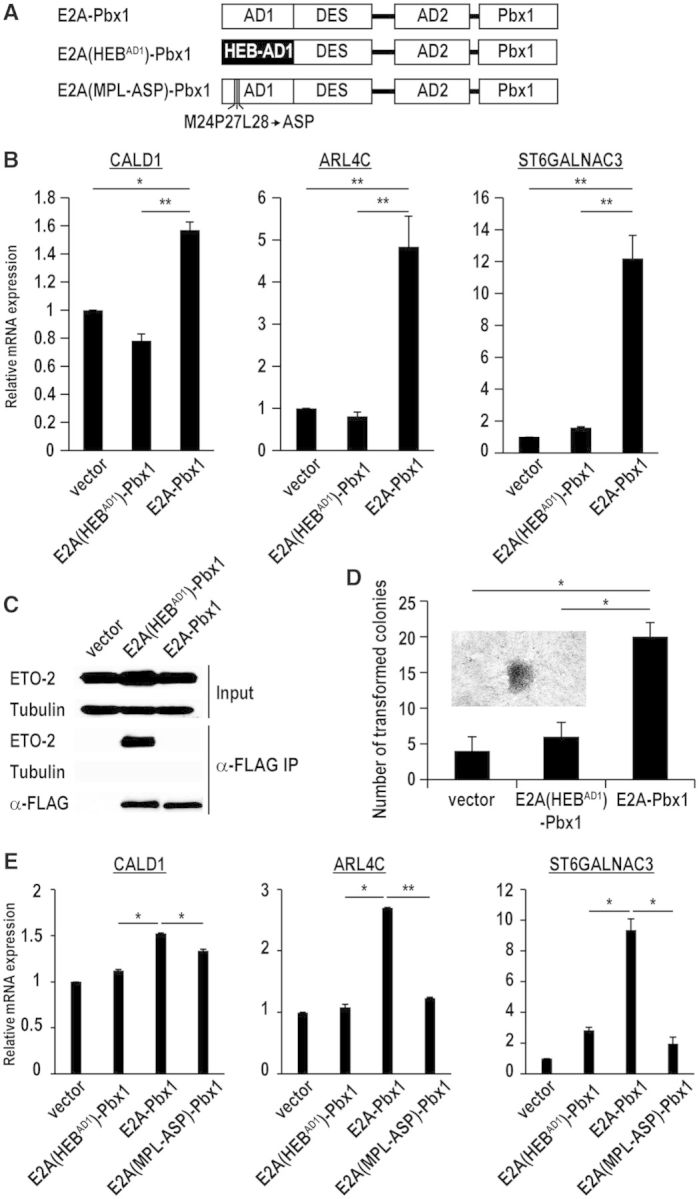
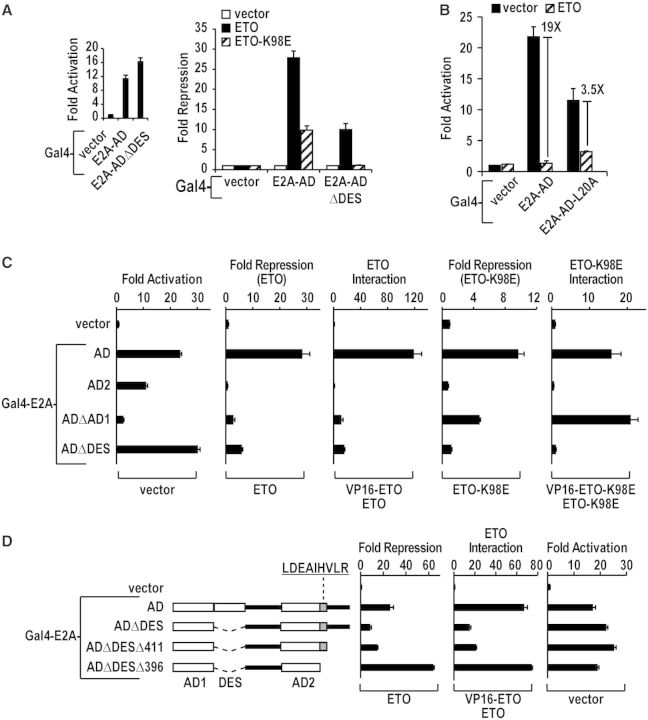
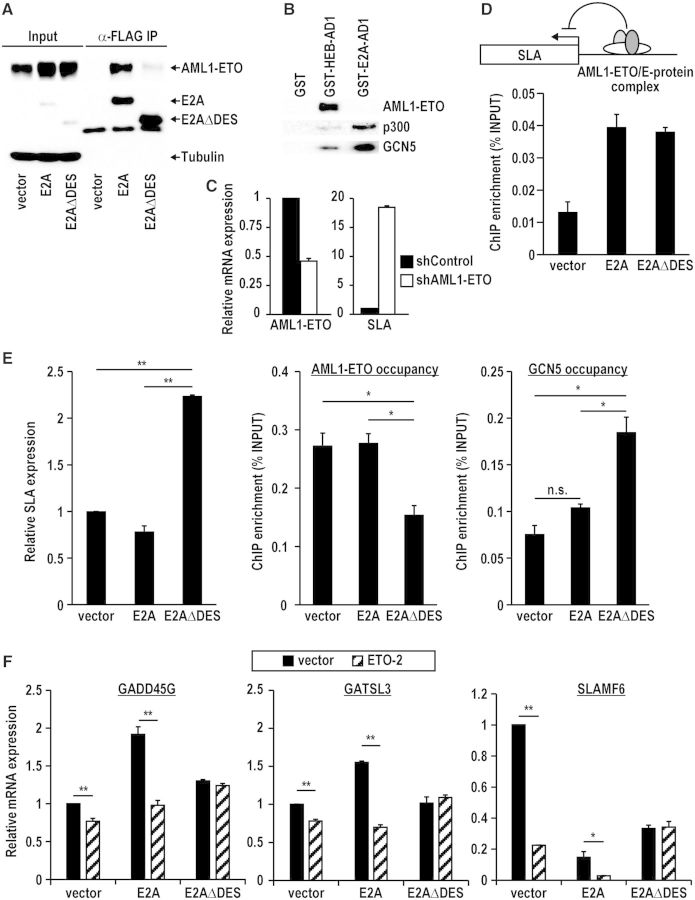
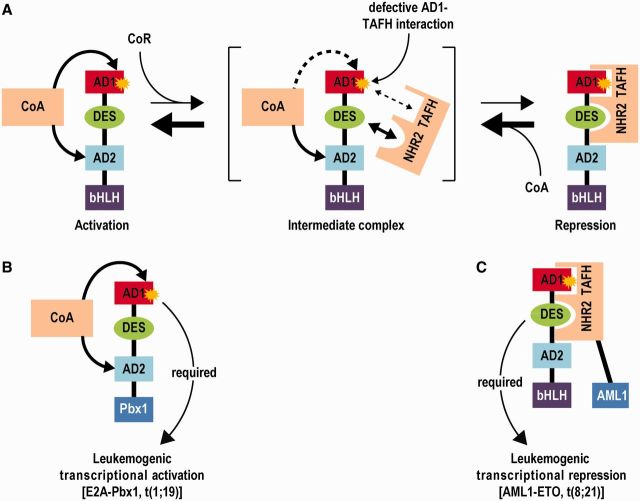
E2A is a member of the E-protein family of transcription factors. Previous studies have reported context-dependent regulation of E2A-dependent transcription. For example, whereas the E2A portion of the E2A-Pbx1 leukemia fusion protein mediates robust transcriptional activation in t(1;19) acute lymphoblastic leukemia, the transcriptional activity of wild-type E2A is silenced by high levels of corepressors, such as the AML1-ETO fusion protein in t(8;21) acute myeloid leukemia and ETO-2 in hematopoietic cells. Here, we show that, unlike the HEB E-protein, the activation domain 1 (AD1) of E2A has specifically reduced corepressor interaction due to E2A-specific amino acid changes in the p300/CBP and ETO target motif. Replacing E2A-AD1 with HEB-AD1 abolished the ability of E2A-Pbx1 to activate target genes and to induce cell transformation. On the other hand, the weak E2A-AD1-corepressor interaction imposes a critical importance on another ETO-interacting domain, downstream ETO-interacting sequence (DES), for corepressor-mediated repression. Deletion of DES abrogates silencing of E2A activity by AML1-ETO in t(8;21) leukemia cells or by ETO-2 in normal hematopoietic cells. Our results reveal an E2A-specific mechanism important for its context-dependent activation and repression function, and provide the first evidence for the differential involvement of E2A-corepressor interactions in distinct leukemogenic pathways.
Figures
Similar articles
-
Multivalent binding of the ETO corepressor to E proteins facilitates dual repression controls targeting chromatin and the basal transcription machinery.Mol Cell Biol. 2009 May;29(10):2644-57. doi: 10.1128/MCB.00073-09. Epub 2009 Mar 16. Mol Cell Biol. 2009. PMID: 19289505 Free PMC article.
-
Critical role for a single leucine residue in leukemia induction by E2A-PBX1.Mol Cell Biol. 2006 Sep;26(17):6442-52. doi: 10.1128/MCB.02025-05. Mol Cell Biol. 2006. PMID: 16914730 Free PMC article.
-
ETO, but not leukemogenic fusion protein AML1/ETO, augments RBP-Jkappa/SHARP-mediated repression of notch target genes.Mol Cell Biol. 2008 May;28(10):3502-12. doi: 10.1128/MCB.01966-07. Epub 2008 Mar 10. Mol Cell Biol. 2008. PMID: 18332109 Free PMC article.
-
ETO interacting proteins.Oncogene. 2004 May 24;23(24):4270-4. doi: 10.1038/sj.onc.1207674. Oncogene. 2004. PMID: 15156183 Review.
-
Molecular targeting of aberrant transcription factors in leukemia: strategies for RUNX1/ETO.Curr Drug Targets. 2010 Sep;11(9):1181-91. doi: 10.2174/138945010792006744. Curr Drug Targets. 2010. PMID: 20583973 Review.
Cited by
-
Computational Modeling of Gene-Specific Transcriptional Repression, Activation and Chromatin Interactions in Leukemogenesis by LASSO-Regularized Logistic Regression.IEEE/ACM Trans Comput Biol Bioinform. 2021 Nov-Dec;18(6):2109-2122. doi: 10.1109/TCBB.2021.3078128. Epub 2021 Dec 8. IEEE/ACM Trans Comput Biol Bioinform. 2021. PMID: 33961561 Free PMC article.
-
Regulation of mammalian transcription and splicing by Nuclear RNAi.Nucleic Acids Res. 2016 Jan 29;44(2):524-37. doi: 10.1093/nar/gkv1305. Epub 2015 Nov 26. Nucleic Acids Res. 2016. PMID: 26612865 Free PMC article. Review.
-
Signaling networks controlling ID and E protein activity in T cell differentiation and function.Front Immunol. 2022 Aug 2;13:964581. doi: 10.3389/fimmu.2022.964581. eCollection 2022. Front Immunol. 2022. PMID: 35983065 Free PMC article. Review.
-
Myeloid translocation gene CBFA2T3 directs a relapse gene program and determines patient-specific outcomes in AML.Blood Adv. 2019 May 14;3(9):1379-1393. doi: 10.1182/bloodadvances.2018028514. Blood Adv. 2019. PMID: 31040112 Free PMC article.
-
New insights into transcriptional and leukemogenic mechanisms of AML1-ETO and E2A fusion proteins.Front Biol (Beijing). 2016 Aug;11(4):285-304. doi: 10.1007/s11515-016-1415-1. Epub 2016 Sep 3. Front Biol (Beijing). 2016. PMID: 28261265 Free PMC article.
References
-
- Murre C, Bain G, van Dijk MA, Engel I, Furnari BA, Massari ME, Matthews JR, Quong MW, Rivera RR, Stuiver MH. Structure and function of helix-loop-helix proteins. Biochim. Biophys. Acta. 1994;1218:129–135. - PubMed
-
- Kee BL, Quong MW, Murre C. E2A proteins: essential regulators at multiple stages of B-cell development. Immunol. Rev. 2000;175:138–149. - PubMed
-
- Bain G, Maandag EC, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BC, Krop I, Schlissel MS, Feeney AJ, van Roon M, et al. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. - PubMed
-
- Zhuang Y, Soriano P, Weintraub H. The helix-loop-helix gene E2A is required for B cell formation. Cell. 1994;79:875–884. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
