

Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration
- PMID: 24064337
- PMCID: PMC3900102
- DOI: 10.1093/hmg/ddt468
Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration
Abstract
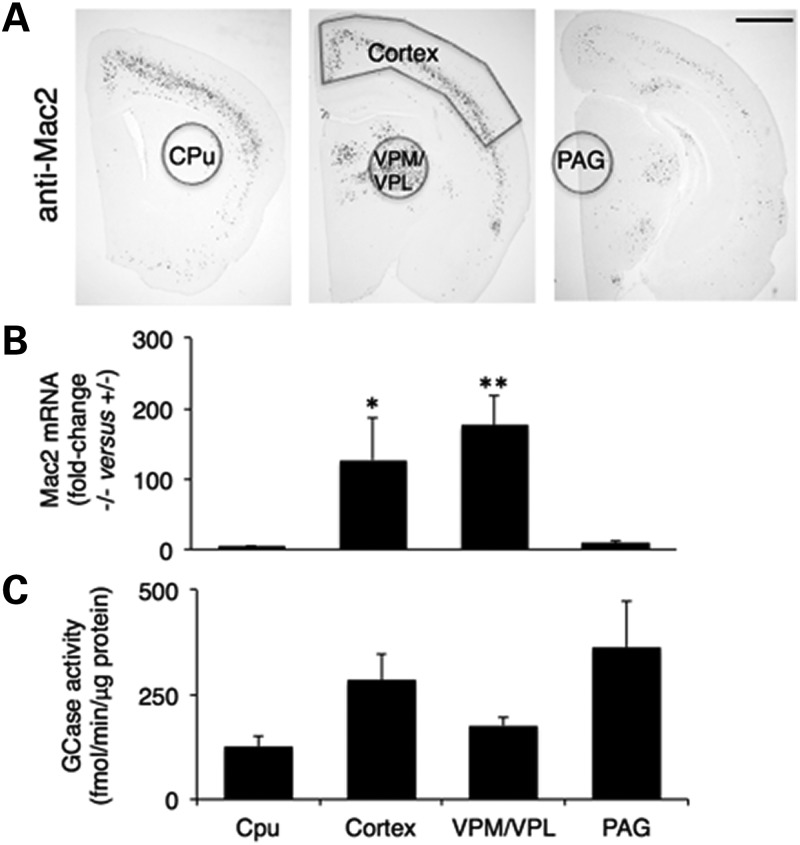
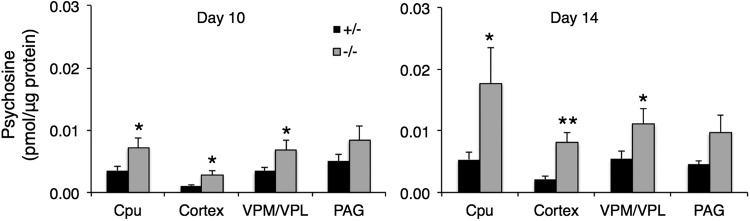
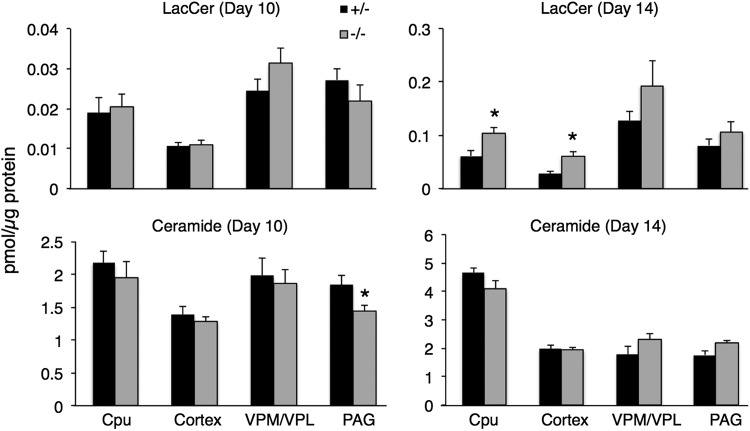
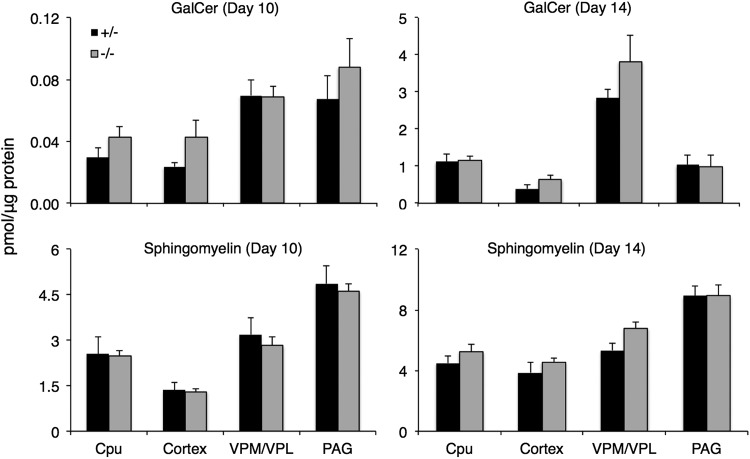
Gaucher disease has recently received wide attention due to the unexpected discovery that it is a genetic risk factor for Parkinson's disease. Gaucher disease is caused by the defective activity of the lysosomal enzyme, glucocerebrosidase (GCase; GBA1), resulting in intracellular accumulation of the glycosphingolipids, glucosylceramide and psychosine. The rare neuronopathic forms of GD (nGD) are characterized by profound neurological impairment and neuronal cell death. We have previously described the progression of neuropathological changes in a mouse model of nGD. We now examine the relationship between glycosphingolipid accumulation and initiation of pathology at two pre-symptomatic stages of the disease in four different brain areas which display differential degrees of susceptibility to GCase deficiency. Liquid chromatography electrospray ionization tandem mass spectrometry demonstrated glucosylceramide and psychosine accumulation in nGD brains prior to the appearance of neuroinflammation, although only glucosylceramide accumulation correlated with neuroinflammation and neuron loss. Levels of other sphingolipids, including the pro-apoptotic lipid, ceramide, were mostly unaltered. Transmission electron microscopy revealed that glucosylceramide accumulation occurs in neurons, mostly in the form of membrane-delimited pseudo-tubules located near the nucleus. Highly disrupted glucosylceramide-storing cells, which are likely degenerating neurons containing massive inclusions, numerous autophagosomes and unique ultrastructural features, were also observed. Together, our results indicate that a certain level of neuronal glucosylceramide storage is required to trigger neuropathological changes in affected brain areas, while other brain areas containing similar glucosylceramide levels are unaltered, presumably because of intrinsic differences in neuronal properties, or in the neuronal environment, between various brain regions.
Figures






References
-
- Farfel-Becker T., Futerman A.H. Cellular pathogenesis in sphingolipid storage disorders: the quest for new therapeutic approaches. Clin. Lipidol. 2010;5:255–265.
-
- Beutler E., Grabowski G.A. Gaucher Disease. In: Scriver C., Beaudet A.L., Valle D., Sly W.S., Childs B., editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. 3. New-York: McGraw-Hill; 2001. pp. 3635–3668.
-
- Mignot C., Doummar D., Maire I., De Villemeur T.B. French Type 2 Gaucher Disease Study Group. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006;28:39–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
