

Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein
- PMID: 24064791
- PMCID: PMC3851721
- DOI: 10.1261/rna.040709.113
Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein
Abstract
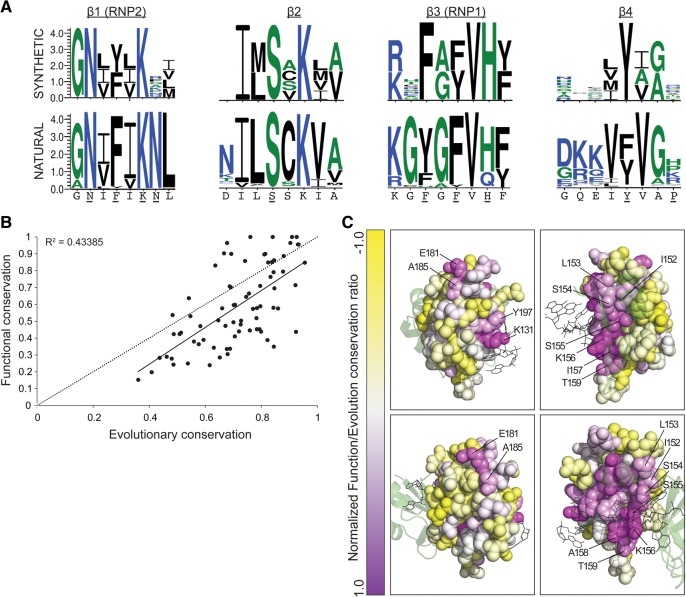
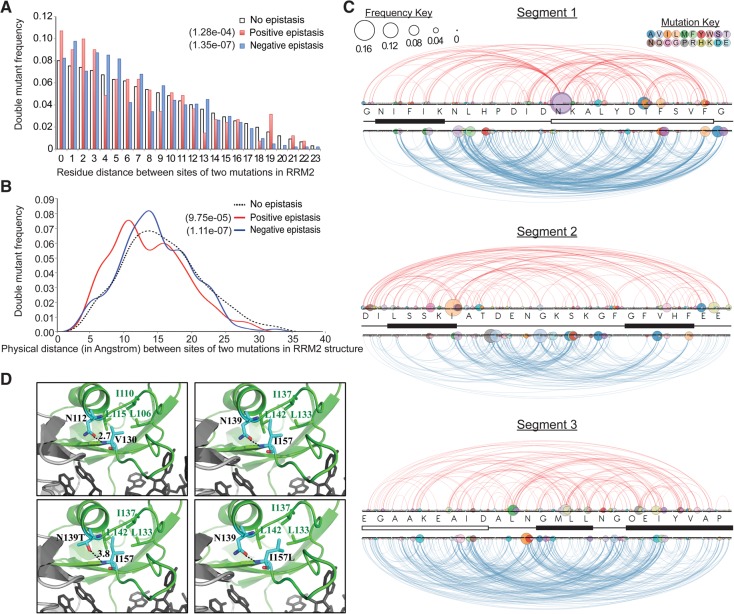
The RNA recognition motif (RRM) is the most common RNA-binding domain in eukaryotes. Differences in RRM sequences dictate, in part, both RNA and protein-binding specificities and affinities. We used a deep mutational scanning approach to study the sequence-function relationship of the RRM2 domain of the Saccharomyces cerevisiae poly(A)-binding protein (Pab1). By scoring the activity of more than 100,000 unique Pab1 variants, including 1246 with single amino acid substitutions, we delineated the mutational constraints on each residue. Clustering of residues with similar mutational patterns reveals three major classes, composed principally of RNA-binding residues, of hydrophobic core residues, and of the remaining residues. The first class also includes a highly conserved residue not involved in RNA binding, G150, which can be mutated to destabilize Pab1. A comparison of the mutational sensitivity of yeast Pab1 residues to their evolutionary conservation reveals that most residues tolerate more substitutions than are present in the natural sequences, although other residues that tolerate fewer substitutions may point to specialized functions in yeast. An analysis of ∼40,000 double mutants indicates a preference for a short distance between two mutations that display an epistatic interaction. As examples of interactions, the mutations N139T, N139S, and I157L suppress other mutations that interfere with RNA binding and protein stability. Overall, this study demonstrates that living cells can be subjected to a single assay to analyze hundreds of thousands of protein variants in parallel.
Keywords: Pab1; RNA recognition motif; RNA-binding protein; epistasis; structure-function analysis.
Figures
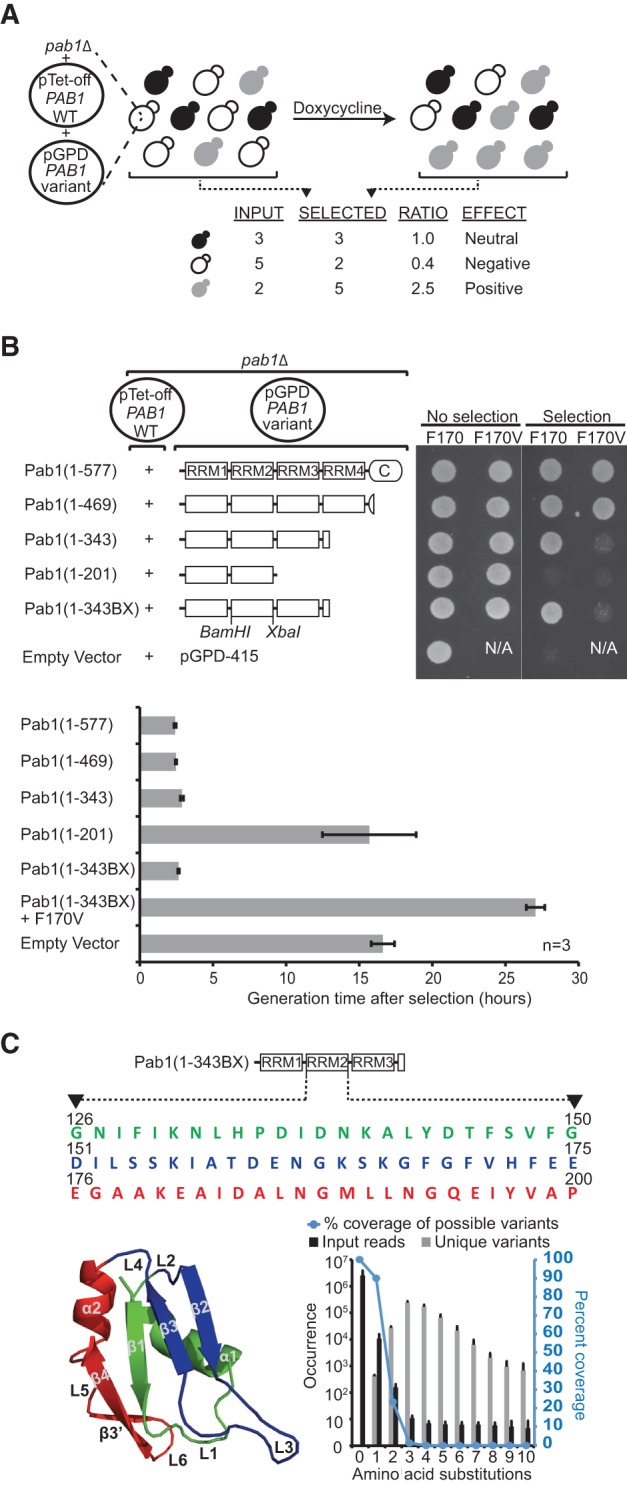
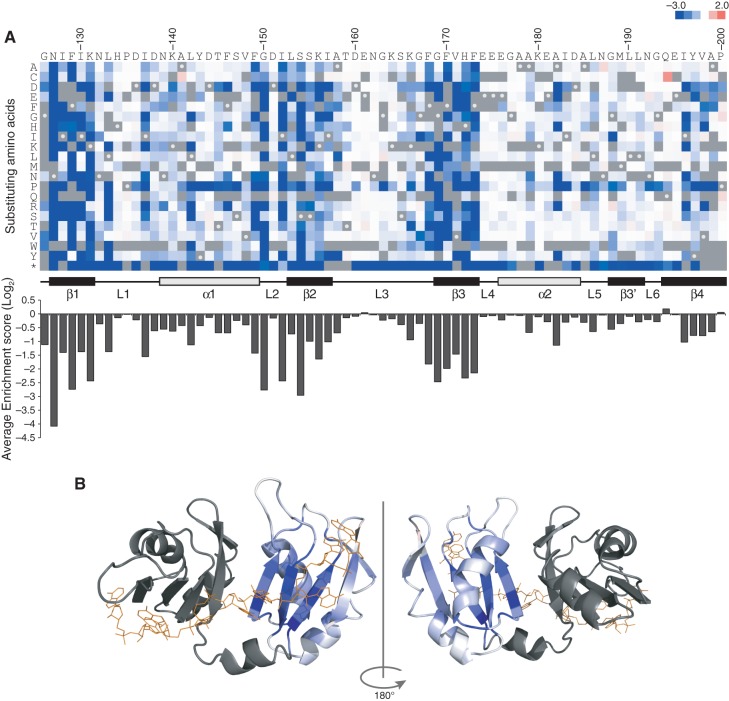
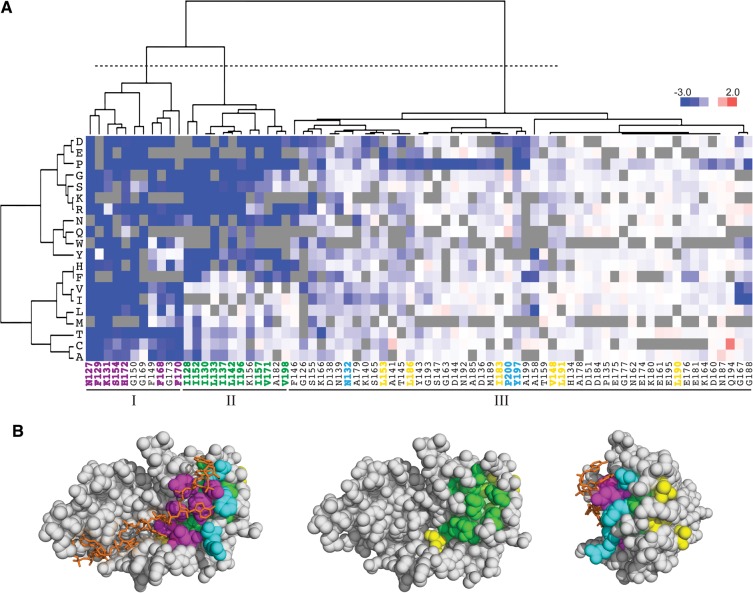
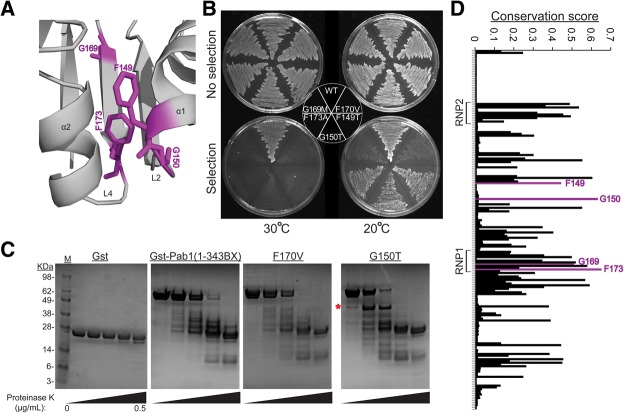
References
-
- Adkar BV, Tripathi A, Sahoo A, Bajaj K, Goswami D, Chakrabarti P, Swarnkar MK, Gokhale RS, Varadarajan R 2012. Protein model discrimination using mutational sensitivity derived from deep sequencing. Structure 20: 371–381 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous