

How to regulate nonbiological complex drugs (NBCD) and their follow-on versions: points to consider
- PMID: 24065600
- PMCID: PMC3889532
- DOI: 10.1208/s12248-013-9533-z
How to regulate nonbiological complex drugs (NBCD) and their follow-on versions: points to consider
Abstract
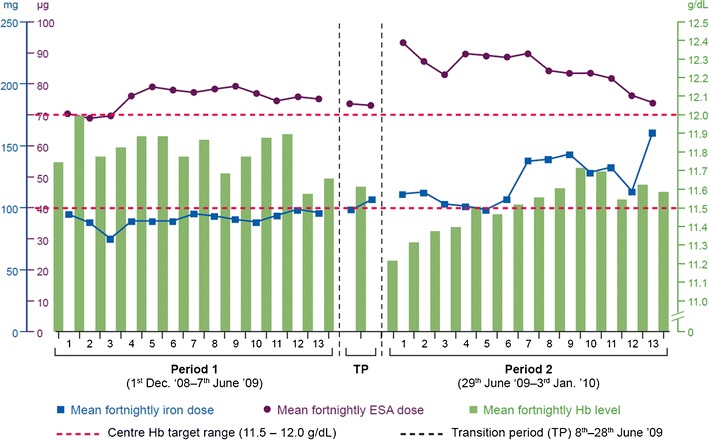
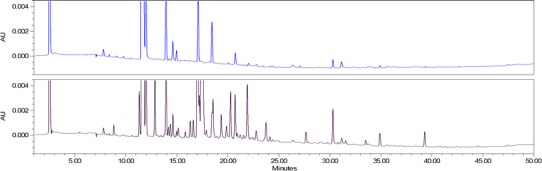
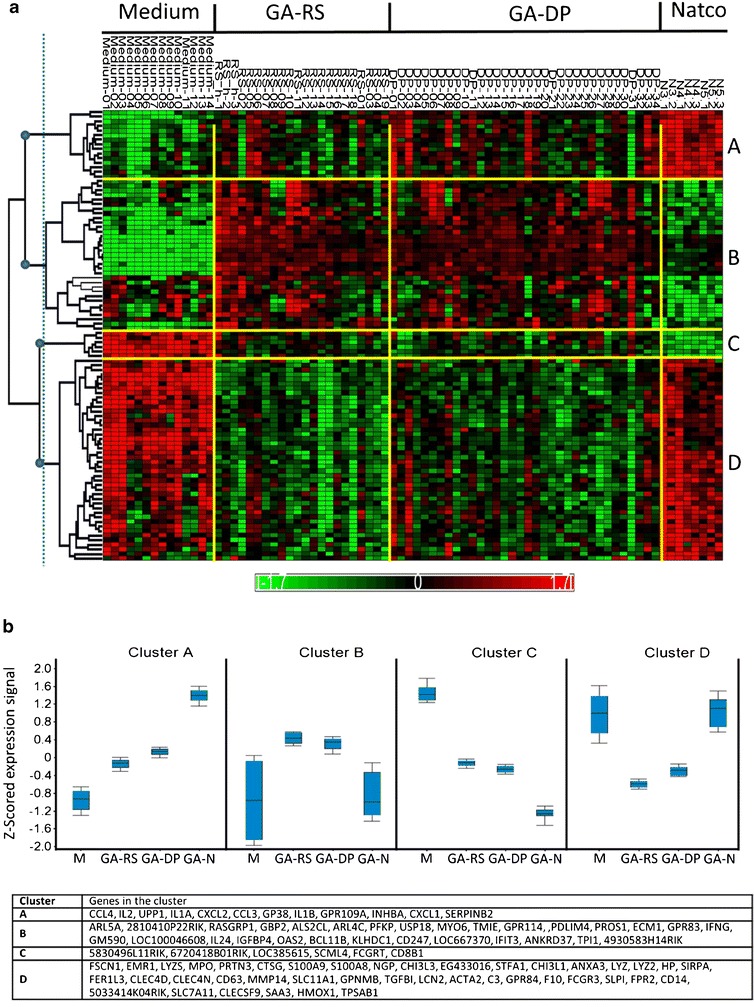
The aim of this critical review is to reach a global consensus regarding the introduction of follow-on versions of nonbiological complex drugs (NBCD). A nonbiological complex drug is a medicinal product, not being a biological medicine, where the active substance is not a homo-molecular structure, but consists of different (closely related and often nanoparticulate) structures that cannot be isolated and fully quantitated, characterized and/or described by state of the art physicochemical analytical means and where the clinical meaning of the differences is not known. The composition, quality and in vivo performance of NBCD are highly dependent on manufacturing processes of both the active ingredient as well as in most cases the formulation. The challenges posed by the development of follow-on versions of NBCD are illustrated in this paper by discussing the 'families' of liposomes, iron-carbohydrate ('iron-sugar') drugs and glatiramoids. It is proposed that the same principles for the marketing authorization of copies of NBCD as for biosimilars be used: the need for animal and/or clinical data and the need to show similarity in quality, safety and efficacy. The regulatory approach of NBCD will have to take into consideration the specific characteristics of the drugs, their formulation and manufacturing process and the resulting critical attributes to achieve their desired quality, safety and efficacy. As with the biosimilars, for the NBCD product, family-specific methods should be evaluated and applied where scientifically proven, including sophisticated quality methods, pharmacodynamic markers and animal models. Concerning substitution and interchangeability of NBCD, it is also advisable to take biosimilars as an example, i.e. (1) substitution without the involvement of a healthcare professional should be discouraged to ensure traceability of the treatment of individual patients, (2) keep an individual patient on a specific treatment if the patient is doing well and only switch if unavoidable and (3) monitor the safety and efficacy of the new product if switching occurs.
Figures
References
-
- Schellekens H, Klinger E, Mühlebach S, Brin JF, Storm G, Crommelin DJA. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59:176–83. - PubMed
-
- Holloway C, Mueller-Berghaus J, Lima BS, Lee SL, Wyatt JS, Nicholas JM, et al. Scientific considerations for complex drugs in light of established and emerging regulatory guidance. Ann N Y Acad Sci. 2012;1276:26–36. - PubMed
-
- Mamidi RNVS, Weng S, Stellar S, Wang C, Yu N, Huang T, et al. Pharmacokinetics, efficacy and toxicity of different pegylated liposomal doxorubicin formulations in preclinical models: is a conventional bioequivalence approach sufficient to ensure therapeutic equivalence of pegylated liposomal doxorubicin products? Cancer Chemother Pharmacol. 2010;66:1173–84. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
