

Type I restriction enzymes and their relatives
- PMID: 24068554
- PMCID: PMC3874165
- DOI: 10.1093/nar/gkt847
Type I restriction enzymes and their relatives
Abstract
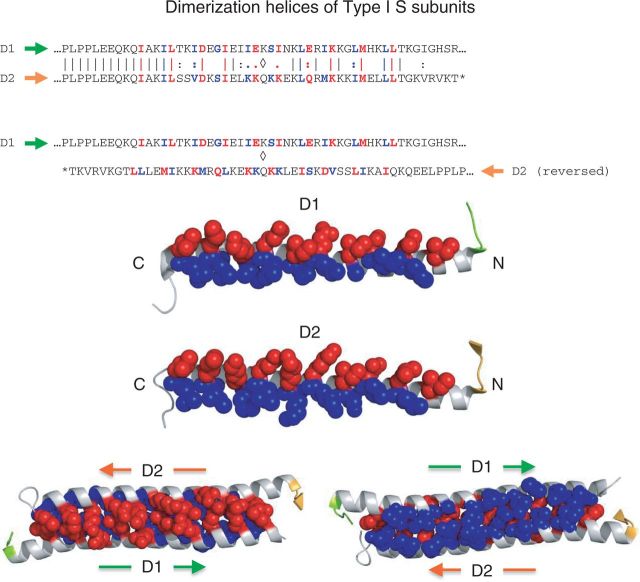
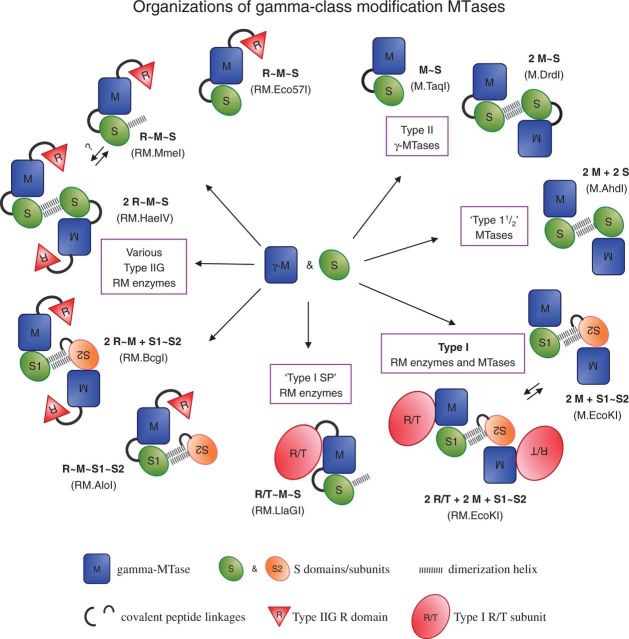
Type I restriction enzymes (REases) are large pentameric proteins with separate restriction (R), methylation (M) and DNA sequence-recognition (S) subunits. They were the first REases to be discovered and purified, but unlike the enormously useful Type II REases, they have yet to find a place in the enzymatic toolbox of molecular biologists. Type I enzymes have been difficult to characterize, but this is changing as genome analysis reveals their genes, and methylome analysis reveals their recognition sequences. Several Type I REases have been studied in detail and what has been learned about them invites greater attention. In this article, we discuss aspects of the biochemistry, biology and regulation of Type I REases, and of the mechanisms that bacteriophages and plasmids have evolved to evade them. Type I REases have a remarkable ability to change sequence specificity by domain shuffling and rearrangements. We summarize the classic experiments and observations that led to this discovery, and we discuss how this ability depends on the modular organizations of the enzymes and of their S subunits. Finally, we describe examples of Type II restriction-modification systems that have features in common with Type I enzymes, with emphasis on the varied Type IIG enzymes.
Figures
References
-
- Dussoix D, Arber W. Host specificity of DNA produced by Escherichia coli. II. Control over acceptance of DNA from infecting phage lambda. J. Mol. Biol. 1962;5:37–49. - PubMed
-
- Arber W, Dussoix D. Host specificity of DNA produced by Escherichia coli. I. Host controlled modification of bacteriophage lambda. J. Mol. Biol. 1962;5:18–36. - PubMed
-
- Anderson ES, Felix A. Variation in Vi-phage II of Salmonella typhi. Nature. 1952;170:492–494. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
