

Network signatures of survival in glioblastoma multiforme
- PMID: 24068912
- PMCID: PMC3777929
- DOI: 10.1371/journal.pcbi.1003237
Network signatures of survival in glioblastoma multiforme
Abstract
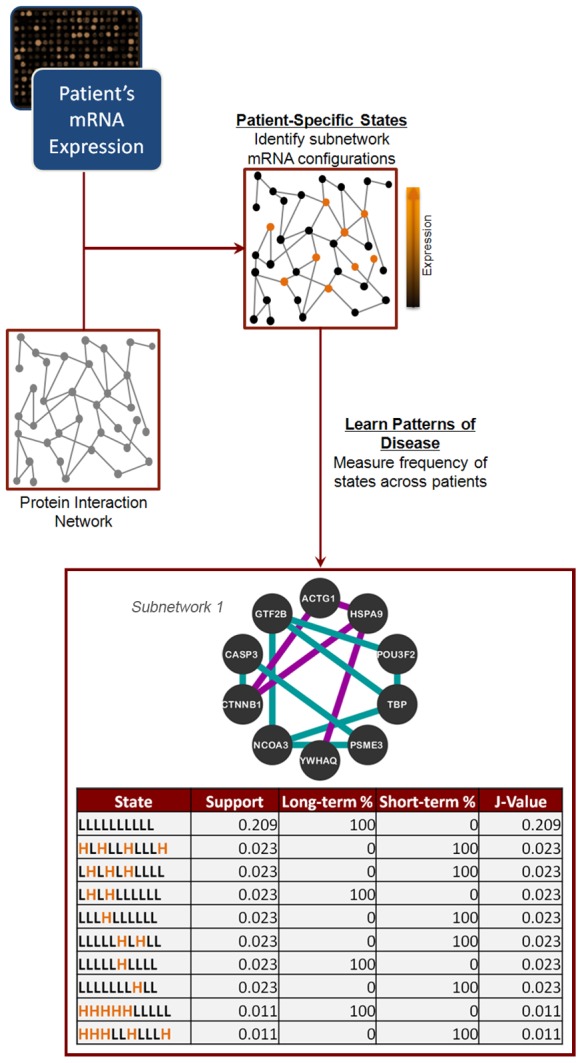
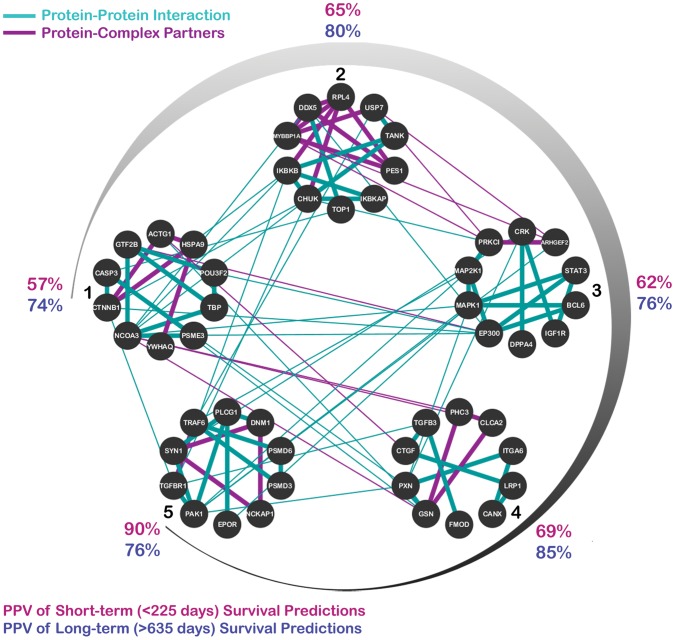
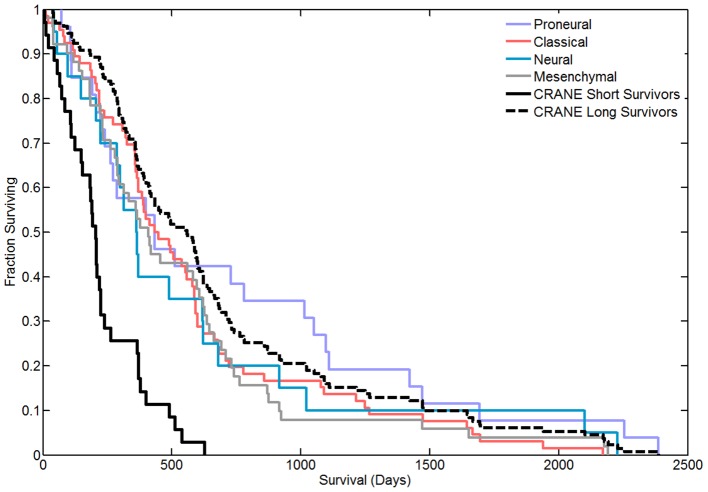
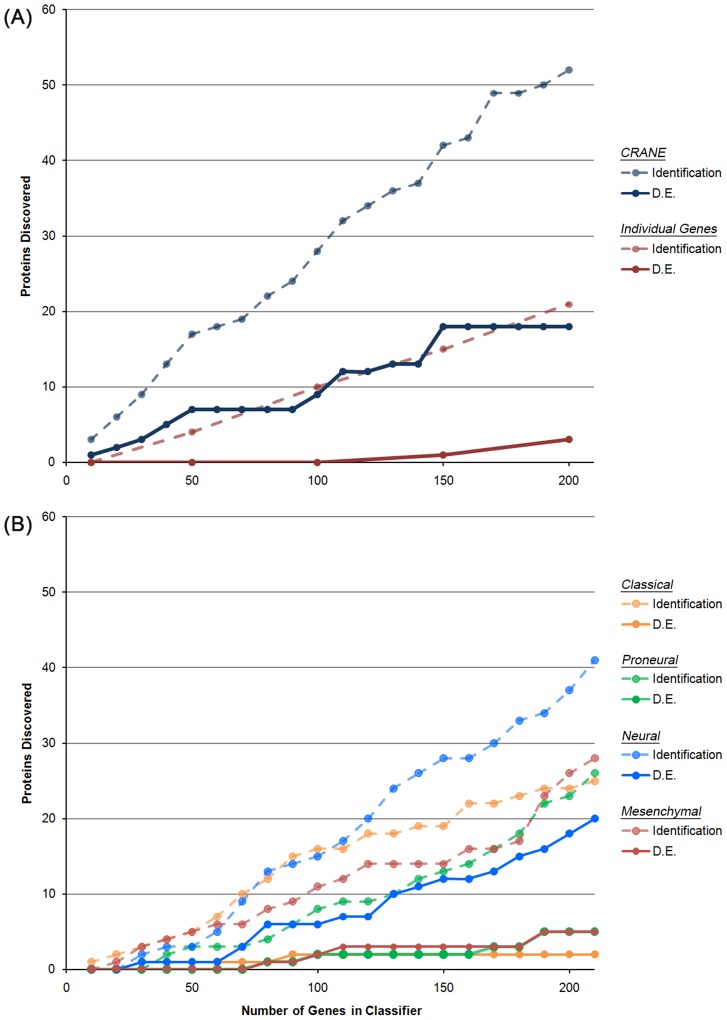
To determine a molecular basis for prognostic differences in glioblastoma multiforme (GBM), we employed a combinatorial network analysis framework to exhaustively search for molecular patterns in protein-protein interaction (PPI) networks. We identified a dysregulated molecular signature distinguishing short-term (survival<225 days) from long-term (survival>635 days) survivors of GBM using whole genome expression data from The Cancer Genome Atlas (TCGA). A 50-gene subnetwork signature achieved 80% prediction accuracy when tested against an independent gene expression dataset. Functional annotations for the subnetwork signature included "protein kinase cascade," "IκB kinase/NFκB cascade," and "regulation of programmed cell death" - all of which were not significant in signatures of existing subtypes. Finally, we used label-free proteomics to examine how our subnetwork signature predicted protein level expression differences in an independent GBM cohort of 16 patients. We found that the genes discovered using network biology had a higher probability of dysregulated protein expression than either genes exhibiting individual differential expression or genes derived from known GBM subtypes. In particular, the long-term survivor subtype was characterized by increased protein expression of DNM1 and MAPK1 and decreased expression of HSPA9, PSMD3, and CANX. Overall, we demonstrate that the combinatorial analysis of gene expression data constrained by PPIs outlines an approach for the discovery of robust and translatable molecular signatures in GBM.
Conflict of interest statement
MRC, SAC, and MK have competing interests in the software CRANE.
Figures
References
-
- Rich JN, Hans C, Jones B, Iversen ES, McLendon RE, et al. (2005) Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res 65: 4051–4058. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
