

Cross-link guided molecular modeling with ROSETTA
- PMID: 24069194
- PMCID: PMC3775805
- DOI: 10.1371/journal.pone.0073411
Cross-link guided molecular modeling with ROSETTA
Abstract
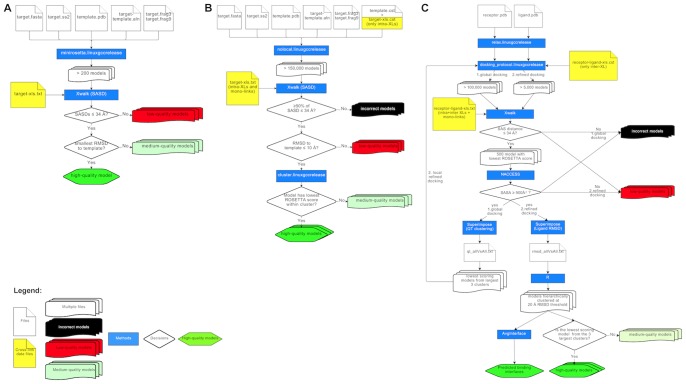
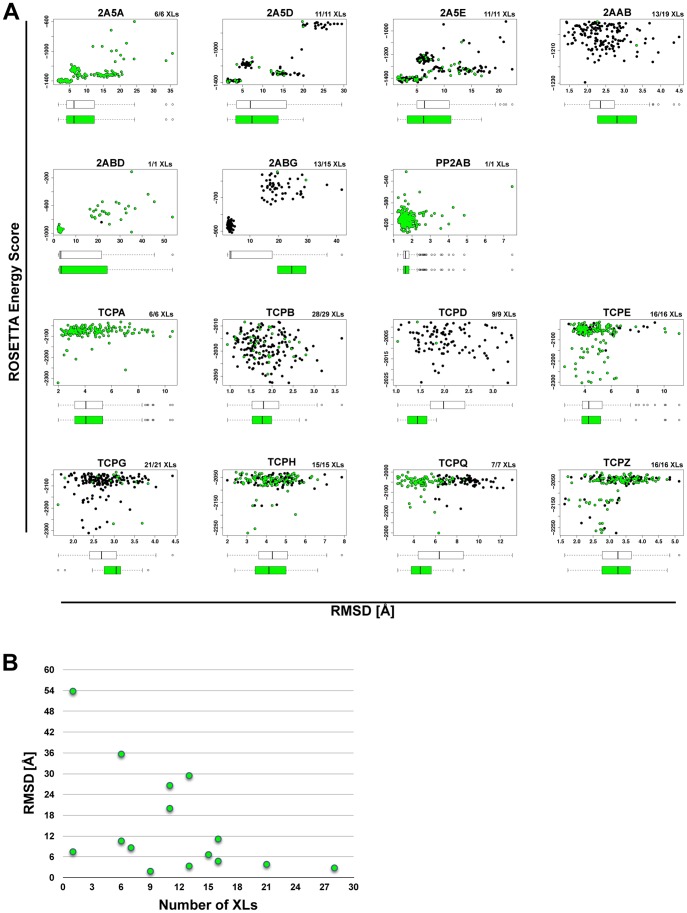
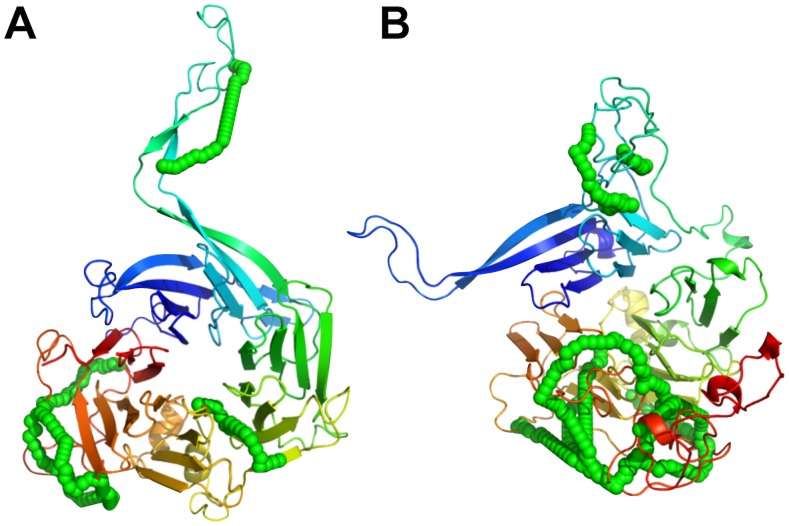
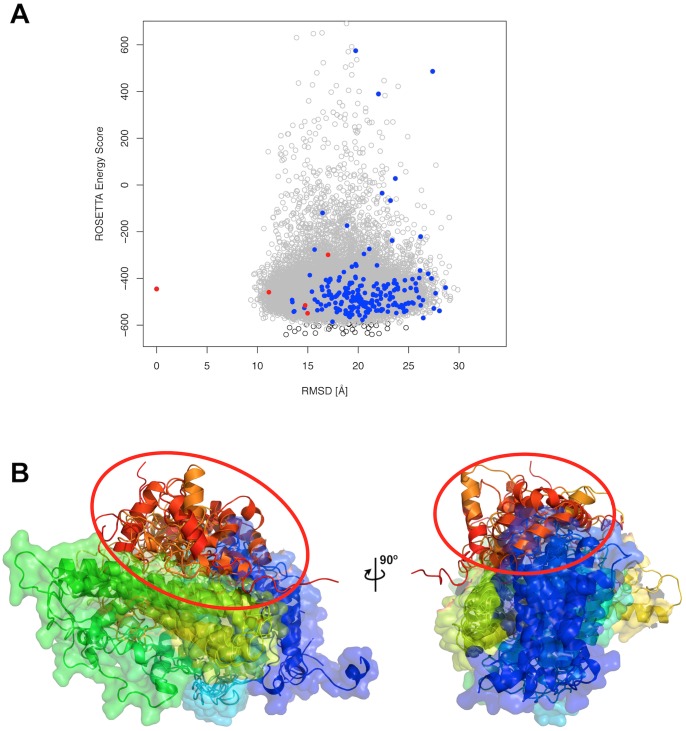
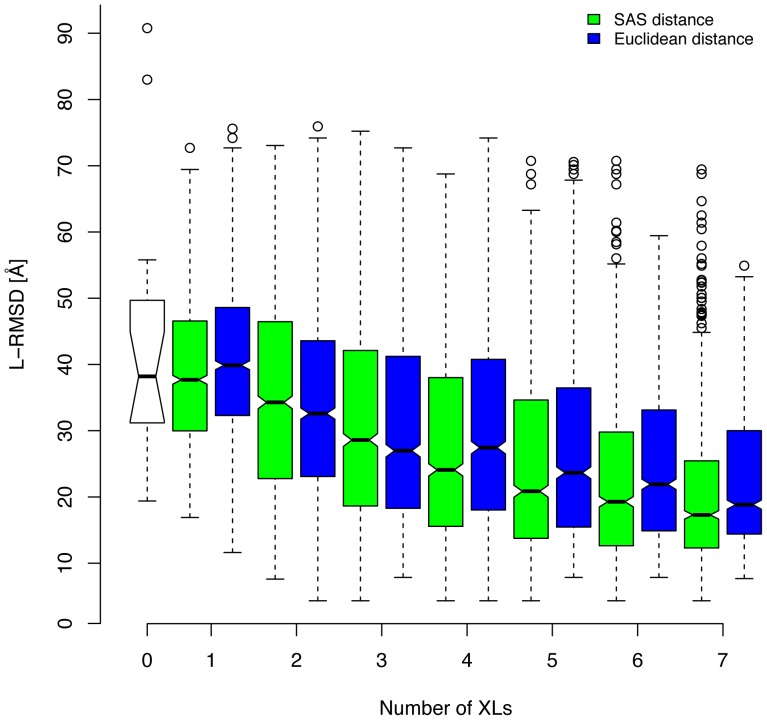
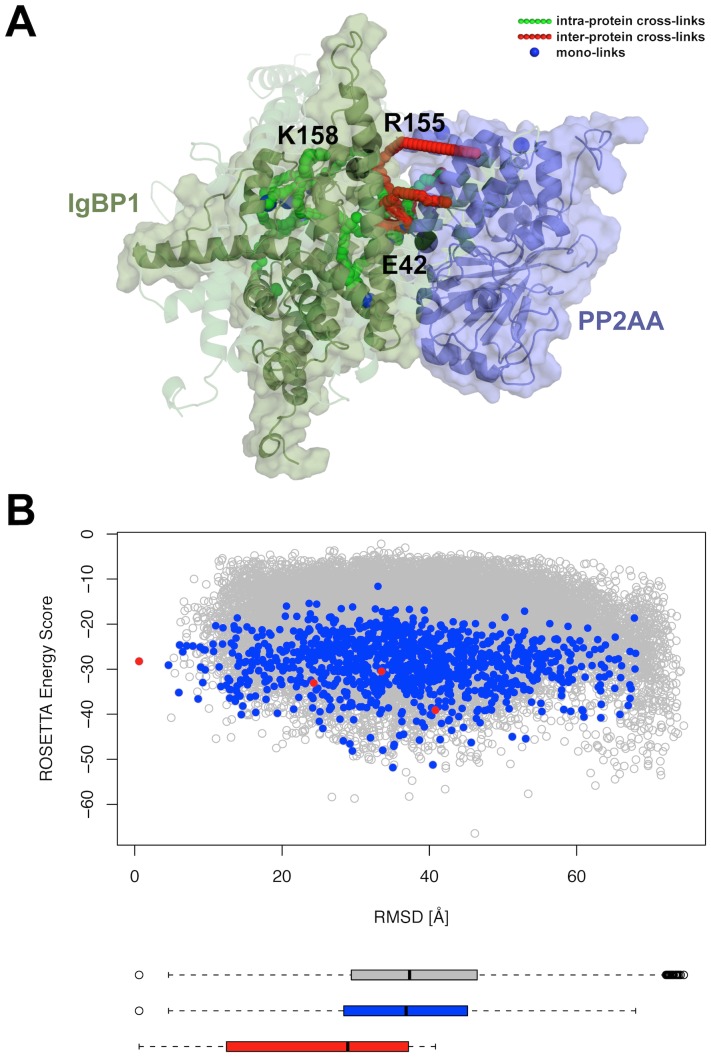
Chemical cross-links identified by mass spectrometry generate distance restraints that reveal low-resolution structural information on proteins and protein complexes. The technology to reliably generate such data has become mature and robust enough to shift the focus to the question of how these distance restraints can be best integrated into molecular modeling calculations. Here, we introduce three workflows for incorporating distance restraints generated by chemical cross-linking and mass spectrometry into ROSETTA protocols for comparative and de novo modeling and protein-protein docking. We demonstrate that the cross-link validation and visualization software Xwalk facilitates successful cross-link data integration. Besides the protocols we introduce XLdb, a database of chemical cross-links from 14 different publications with 506 intra-protein and 62 inter-protein cross-links, where each cross-link can be mapped on an experimental structure from the Protein Data Bank. Finally, we demonstrate on a protein-protein docking reference data set the impact of virtual cross-links on protein docking calculations and show that an inter-protein cross-link can reduce on average the RMSD of a docking prediction by 5.0 Å. The methods and results presented here provide guidelines for the effective integration of chemical cross-link data in molecular modeling calculations and should advance the structural analysis of particularly large and transient protein complexes via hybrid structural biology methods.
Conflict of interest statement
Figures
References
-
- Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, et al... (2012) Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature. doi:10.1038/nature11503. - DOI - PMC - PubMed
-
- Wang X, Wei X, Thijssen B, Das J, Lipkin SM, et al. (2012) Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat Biotechnol 30: 159–164 doi:10.1038/nbt.2106 - DOI - PMC - PubMed
-
- Edwards A (2009) Large-Scale Structural Biology of the Human Proteome. Annu Rev Biochem 78: 541–568 doi:10.1146/annurev.biochem.78.070907.103305 - DOI - PubMed
-
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797. - PubMed
-
- Mosca R, Pons C, Fernández-Recio J, Aloy P (2009) Pushing Structural Information into the Yeast Interactome by High-Throughput Protein Docking Experiments. PLoS Comput Biol 5: e1000490 doi:10.1371/journal.pcbi.1000490 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
