

The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities
- PMID: 24072174
- PMCID: PMC3949163
- DOI: 10.1097/FJC.0000000000000003
The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities
Abstract
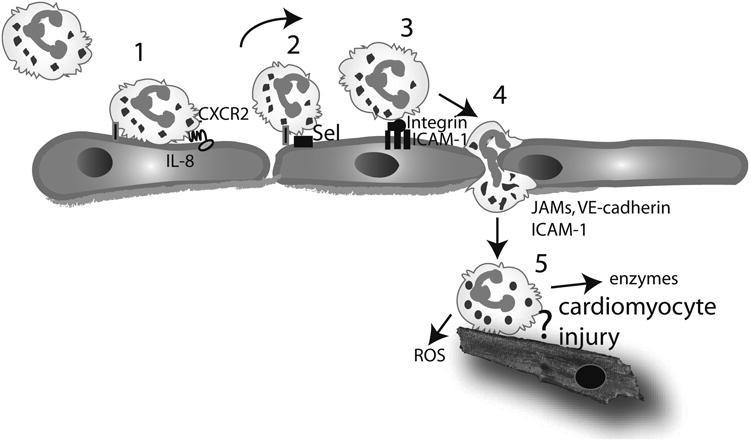
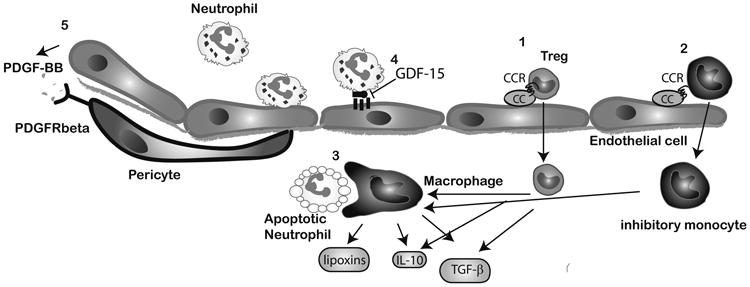
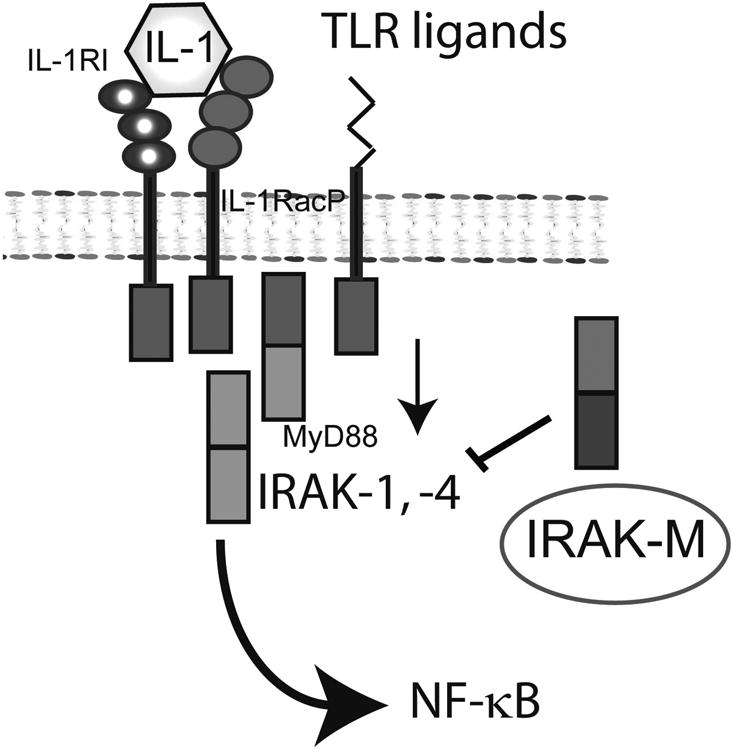
Extensive necrosis of ischemic cardiomyocytes in the infarcted myocardium activates the innate immune response triggering an intense inflammatory reaction. Release of danger signals from dying cells and damaged matrix activates the complement cascade and stimulates Toll-like receptor/interleukin-1 signaling, resulting in the activation of the nuclear factor-κB system and induction of chemokines, cytokines, and adhesion molecules. Subsequent infiltration of the infarct with neutrophils and mononuclear cells serves to clear the wound from dead cells and matrix debris, while stimulating reparative pathways. In addition to its role in repair of the infarcted heart and formation of a scar, the immune system is also involved in adverse remodeling of the infarcted ventricle. Overactive immune responses and defects in suppression, containment, and resolution of the postinfarction inflammatory reaction accentuate dilative remodeling in experimental models and may be associated with chamber dilation, systolic dysfunction, and heart failure in patients surviving a myocardial infarction. Interventions targeting the inflammatory response to attenuate adverse remodeling may hold promise in patients with myocardial infarction that exhibit accentuated, prolonged, or dysregulated immune responses to the acute injury.
Figures
References
-
- Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006 Nov-Dec;8(11-12):1907–1939. - PubMed
-
- Entman ML, Michael L, Rossen RD, Dreyer WJ, Anderson DC, Taylor AA, Smith CW. Inflammation in the course of early myocardial ischemia. Faseb J. 1991 Aug;5(11):2529–2537. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
