

Late-onset ornithine carbamoyltransferase deficiency accompanying acute pancreatitis and hyperammonemia
- PMID: 24073003
- PMCID: PMC3773375
- DOI: 10.1155/2013/903546
Late-onset ornithine carbamoyltransferase deficiency accompanying acute pancreatitis and hyperammonemia
Abstract
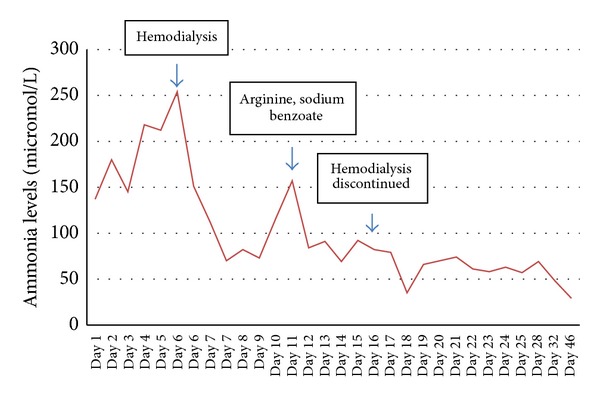
Hyperammonemia related to urea cycle disorders is a rare cause of potentially fatal encephalopathy that is encountered in intensive care units (ICUs). Left undiagnosed, this condition may manifest irreversible neuronal damage. However, timely diagnosis and treatment initiation can be facilitated simply by increased awareness of the ICU staff. Here, we describe a patient with acute severe pancreatitis who developed hyperammonemia and encephalopathy without liver disease. Urea cycle disorder was suspected and hemodialysis was initiated. Following reduction of ammonia levels, subsequent treatment included protein restriction and administration of arginine and sodium benzoate. The patient was discharged to home after 47 days with plasma ammonia within normal range and without neurological symptoms. In clinical care settings, patients with neurological symptoms unexplained by the present illness should be assessed for serum ammonia levels to disclose any urea cycle disorders to initiate timely treatment and improve outcome.
Figures
References
-
- Lanpher BC, Gropman A, Chapman KA, Lichter-Konecki U, Urea Cycle Disorders Consortium, Summar ML. Urea cycle disorders overview. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews [Internet] Seattle, Wash, USA: University of Washington, Seattle; 1993–2003. - PubMed
-
- McCullough BA, Yudkoff M, Batshaw ML, Wilson JM, Raper SE, Tuchman M. Genotype spectrum of ornithine transcarbamylase deficiency: correlation with the clinical and biochemical phenotype. The American Journal of Medical Genetics. 2000;93(4):313–319. - PubMed
-
- Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Advances in Pediatrics. 1996;43:127–170. - PubMed
-
- Caldovic L, Morizono H, Panglao MG, Cheng SF, Packman S, Tuchman M. Null mutations in the N-acetylglutamate synthase gene associated with acute neonatal disease and hyperammonemia. Human Genetics. 2003;112(4):364–368. - PubMed
-
- Yamaguchi S, Brailey LL, Morizono H, Bale AE, Tuchman M. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Human Mutation. 2006;27(7):626–632. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
