

Mutation in MEOX1 gene causes a recessive Klippel-Feil syndrome subtype
- PMID: 24073994
- PMCID: PMC3849745
- DOI: 10.1186/1471-2156-14-95
Mutation in MEOX1 gene causes a recessive Klippel-Feil syndrome subtype
Abstract
Background: Klippel-Feil syndrome (KFS) is characterized by the developmental failure of the cervical spine and has two dominantly inherited subtypes. Affected individuals who are the children of a consanguineous marriage are extremely rare in the medical literature, but the gene responsible for this recessive trait subtype of KFS has recently been reported.
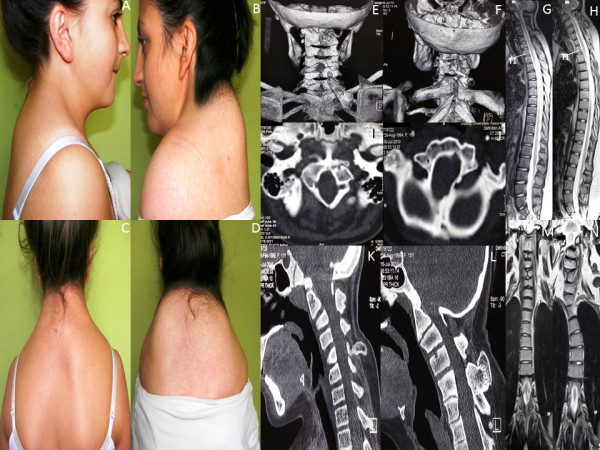
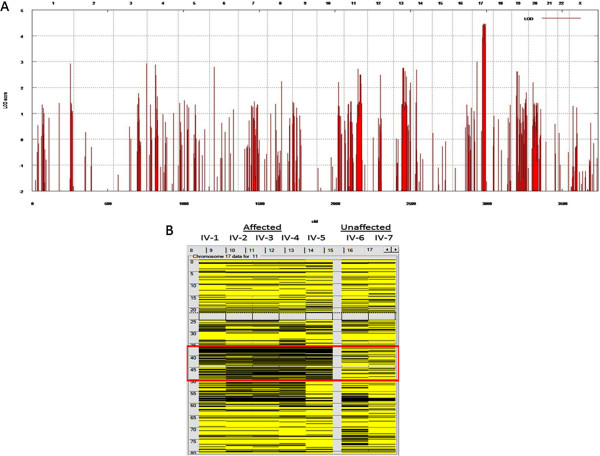
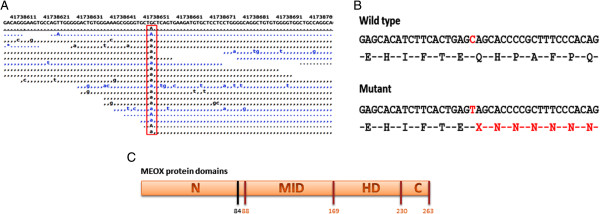
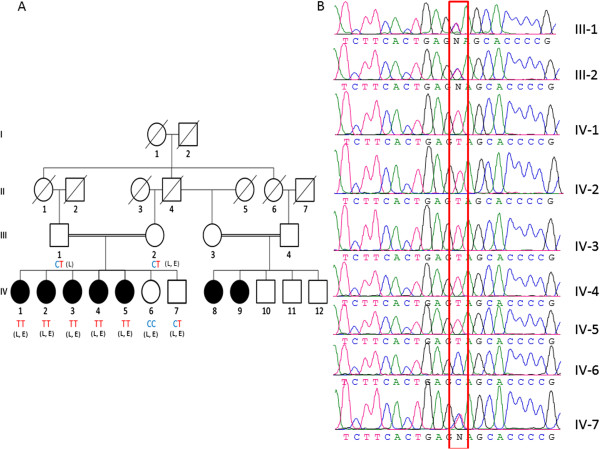
Results: We identified a family with the KFS phenotype in which their parents have a consanguineous marriage. Radiological examinations revealed that they carry fusion defects and numerical abnormalities in the cervical spine, scoliosis, malformations of the cranial base, and Sprengel's deformity. We applied whole genome linkage and whole-exome sequencing analysis to identify the chromosomal locus and gene mutated in this family. Whole genome linkage analysis revealed a significant linkage to chromosome 17q12-q33 with a LOD score of 4.2. Exome sequencing identified the G > A p.Q84X mutation in the MEOX1 gene, which is segregated based on pedigree status. Homozygous MEOX1 mutations have reportedly caused a similar phenotype in knockout mice.
Conclusions: Here, we report a truncating mutation in the MEOX1 gene in a KFS family with an autosomal recessive trait. Together with another recently reported study and the knockout mouse model, our results suggest that mutations in MEOX1 cause a recessive KFS phenotype in humans.
Figures
References
-
- Tracy MR, Dormans JP, Kusumi K. Klippel-Feil syndrome: clinical features and current understanding of etiology. Clin Orthop Relat Res. 2004;424:183–190. - PubMed
-
- Skuntz S, Mankoo B, Nguyen MT, Hustert E, Nakayama A, Tournier-Lasserve E, Wright CV, Pachnis V, Bharti K, Arnheiter H. Lack of the mesodermal homeodomain protein MEOX1 disrupts sclerotome polarity and leads to a remodeling of the cranio-cervical joints of the axial skeleton. Dev Biol. 2009;332(2):383–395. doi: 10.1016/j.ydbio.2009.06.006. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
