

Childhood interstitial lung diseases: an 18-year retrospective analysis
- PMID: 24081995
- PMCID: PMC3784299
- DOI: 10.1542/peds.2013-1780
Childhood interstitial lung diseases: an 18-year retrospective analysis
Abstract
Objective: Childhood interstitial lung diseases (ILD) occur in a variety of clinical contexts. Advances in the understanding of disease pathogenesis and use of standardized terminology have facilitated increased case ascertainment. However, as all studies have been performed at specialized referral centers, the applicability of these findings to general pulmonary practice has been uncertain. The objective of this study was to determine the historical occurrence of childhood ILD to provide information reflecting general pediatric pulmonary practice patterns.
Methods: Childhood ILD cases seen at Vanderbilt Children's Hospital from 1994 to 2011 were retrospectively reviewed and classified according to the current pediatric diffuse lung disease histopathologic classification system.
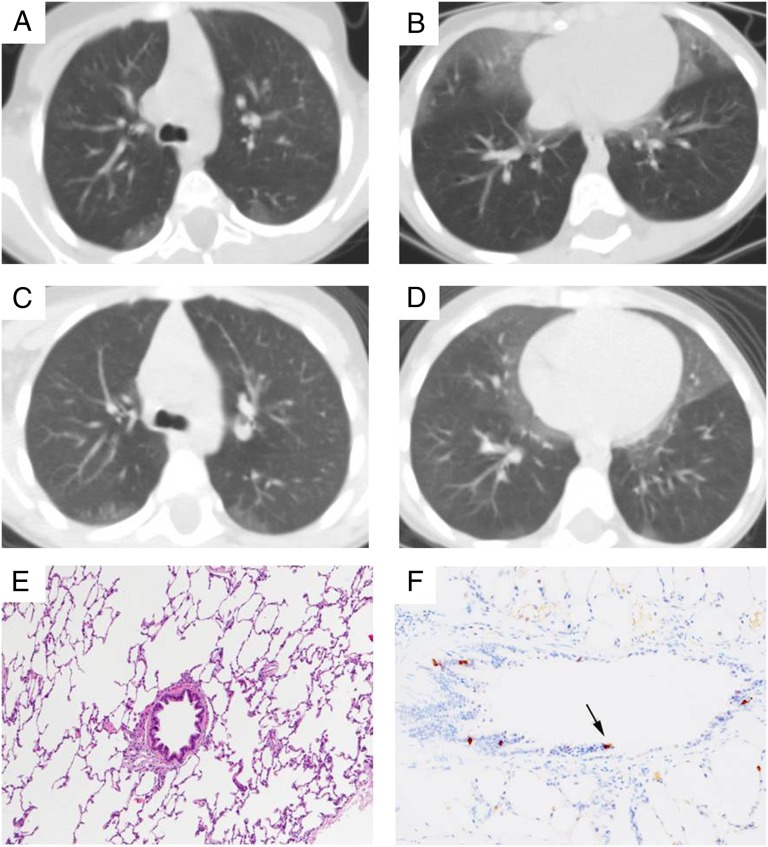
Results: A total of 93 cases were identified, of which 91.4% were classifiable. A total of 68.8% (64/93) of subjects underwent lung biopsy in their evaluations. The largest classification categories were disorders related to systemic disease processes (24.7%), disorders of the immunocompromised host (24.7%), and disorders more prevalent in infancy (22.6%). Eight cases of neuroendocrine cell hyperplasia of infancy (NEHI) were identified, including 5 that were previously unrecognized before this review.
Conclusions: Our findings demonstrate the general scope of childhood ILD and that these cases present within a variety of pediatric subspecialties. Retrospective review was valuable in recognizing more recently described forms of childhood ILD. As a significant portion of cases were classifiable based on clinical, genetic, and/or radiographic criteria, we urge greater consideration to noninvasive diagnostic approaches and suggest modification to the current childhood ILD classification scheme to accommodate the increasing number of cases diagnosed without lung biopsy.
Keywords: childhood lung disease; interstitial lung disease; lung biopsy.
Figures



References
-
- Fan LL, Deterding RR, Langston C. Pediatric interstitial lung disease revisited. Pediatr Pulmonol. 2004;38(5):369–378 - PubMed
-
- Fan LL, Kozinetz CA. Factors influencing survival in children with chronic interstitial lung disease. Am J Respir Crit Care Med. 1997;156(3 pt 1):939–942 - PubMed
-
- Deterding R. Evaluating infants and children with interstitial lung disease. Semin Respir Crit Care Med. 2007;28(3):333–341 - PubMed
-
- Fan LL, Mullen AL, Brugman SM, Inscore SC, Parks DP, White CW. Clinical spectrum of chronic interstitial lung disease in children. J Pediatr. 1992;121(6):867–872 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
