

Discovery of a selective NaV1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models
- PMID: 24082113
- PMCID: PMC3808613
- DOI: 10.1073/pnas.1306285110
Discovery of a selective NaV1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models
Abstract
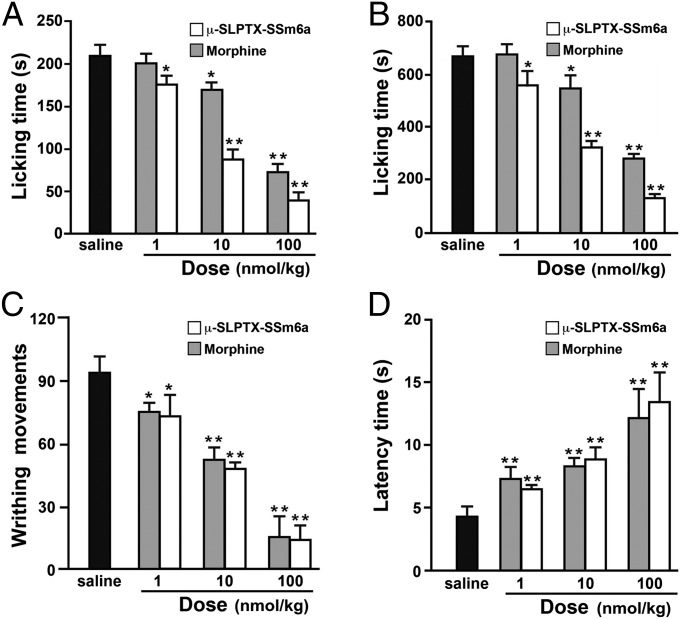
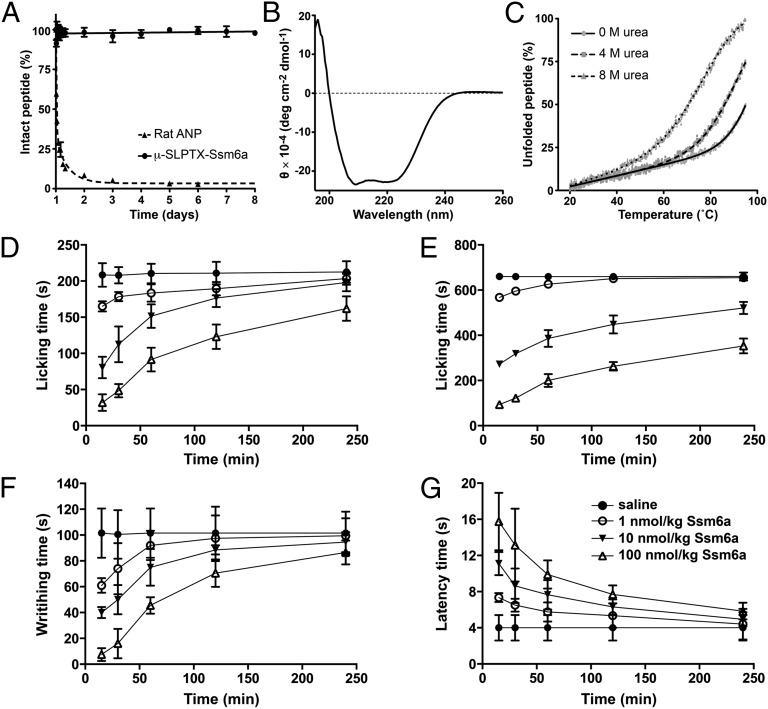
Loss-of-function mutations in the human voltage-gated sodium channel NaV1.7 result in a congenital indifference to pain. Selective inhibitors of NaV1.7 are therefore likely to be powerful analgesics for treating a broad range of pain conditions. Herein we describe the identification of µ-SLPTX-Ssm6a, a unique 46-residue peptide from centipede venom that potently inhibits NaV1.7 with an IC50 of ∼25 nM. µ-SLPTX-Ssm6a has more than 150-fold selectivity for NaV1.7 over all other human NaV subtypes, with the exception of NaV1.2, for which the selectivity is 32-fold. µ-SLPTX-Ssm6a contains three disulfide bonds with a unique connectivity pattern, and it has no significant sequence homology with any previously characterized peptide or protein. µ-SLPTX-Ssm6a proved to be a more potent analgesic than morphine in a rodent model of chemical-induced pain, and it was equipotent with morphine in rodent models of thermal and acid-induced pain. This study establishes µ-SPTX-Ssm6a as a promising lead molecule for the development of novel analgesics targeting NaV1.7, which might be suitable for treating a wide range of human pain pathologies.
Keywords: chronic pain; drug discovery; peptide therapeutic.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
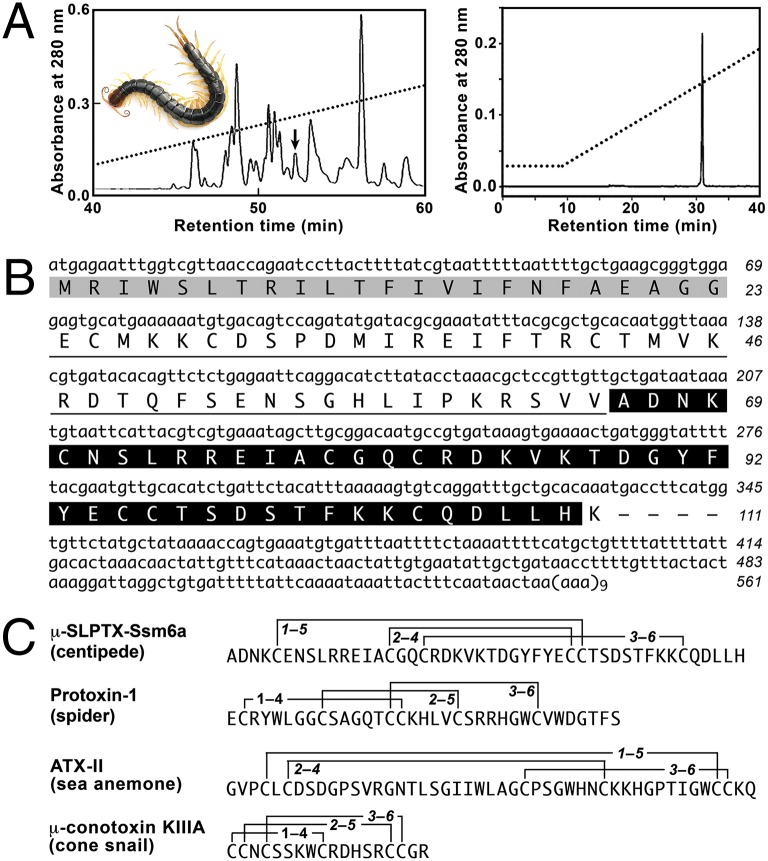
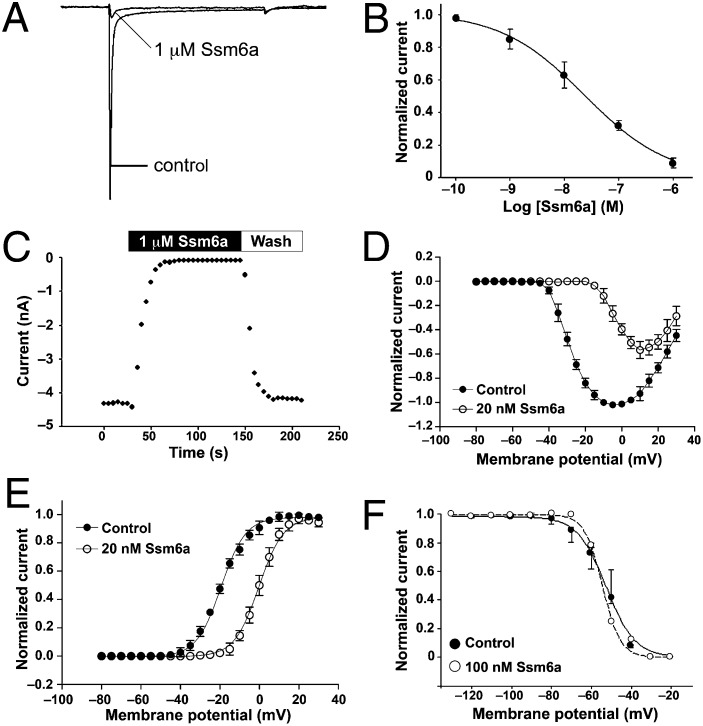
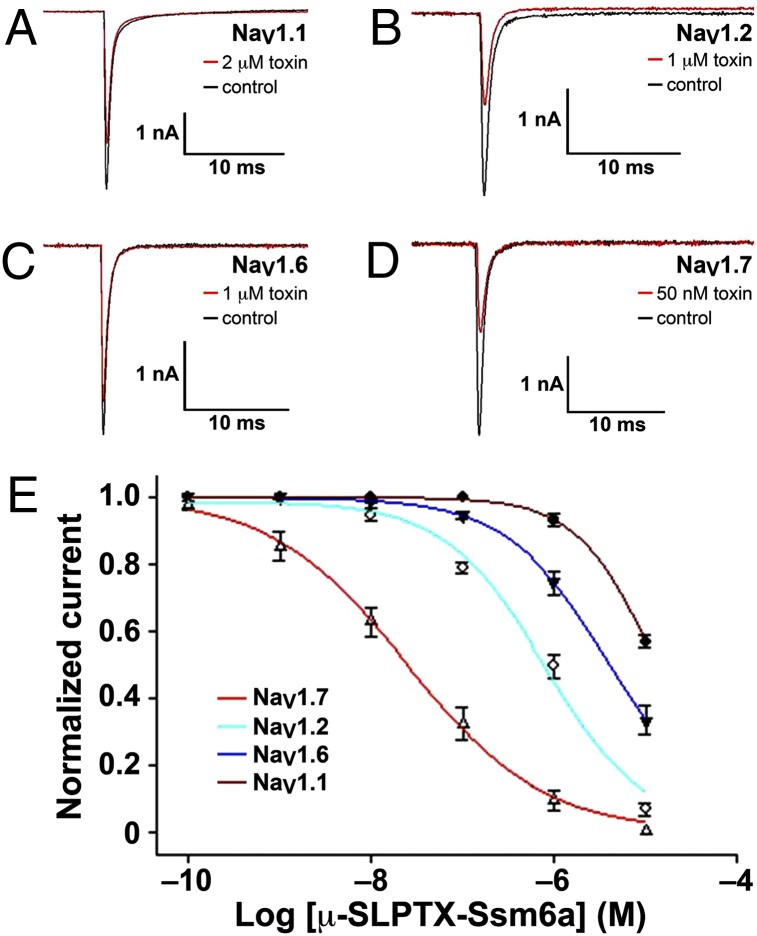
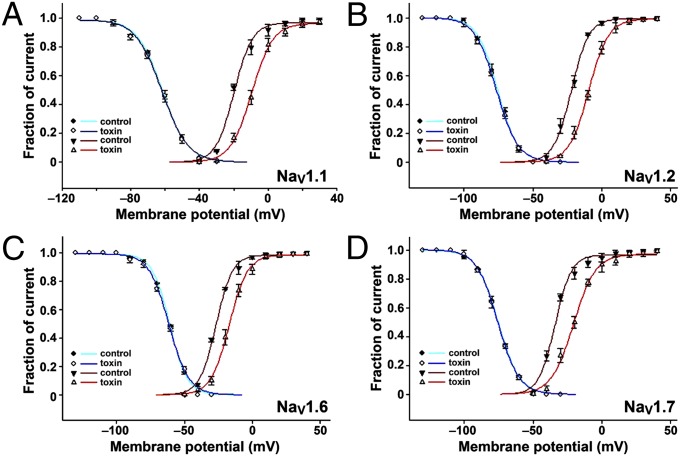
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
