

The cysteine protease cathepsin B is a key drug target and cysteine protease inhibitors are potential therapeutics for traumatic brain injury
- PMID: 24083575
- PMCID: PMC3934599
- DOI: 10.1089/neu.2013.2944
The cysteine protease cathepsin B is a key drug target and cysteine protease inhibitors are potential therapeutics for traumatic brain injury
Abstract
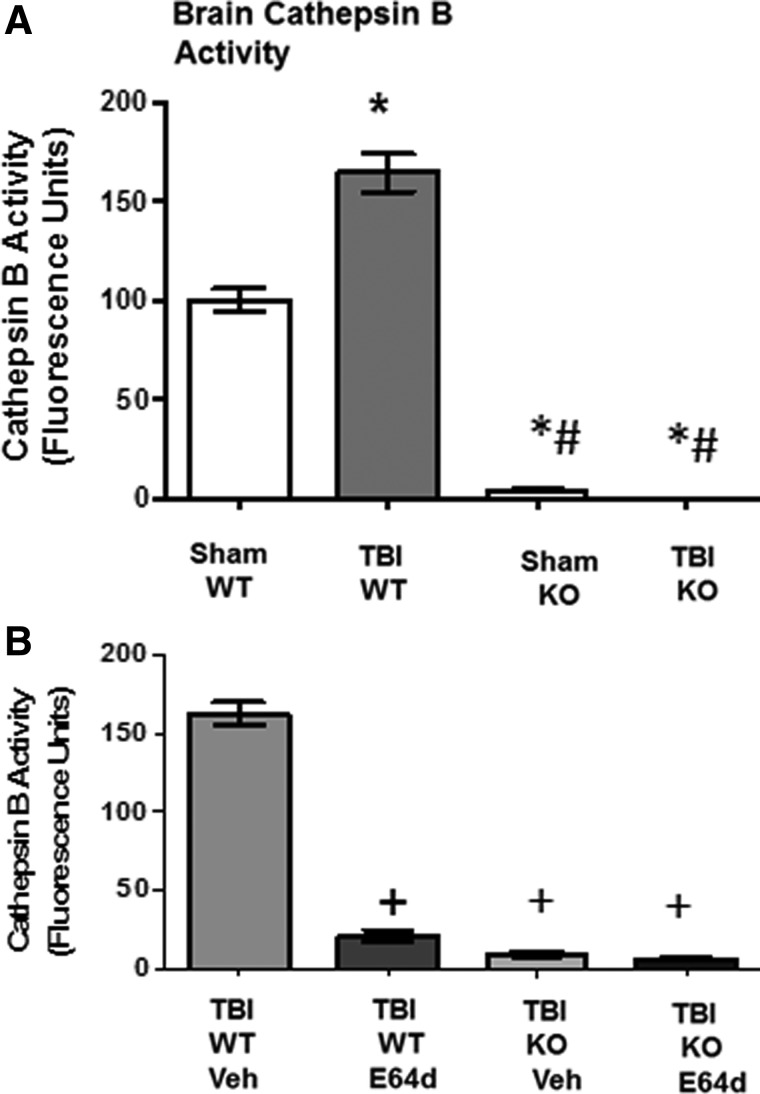
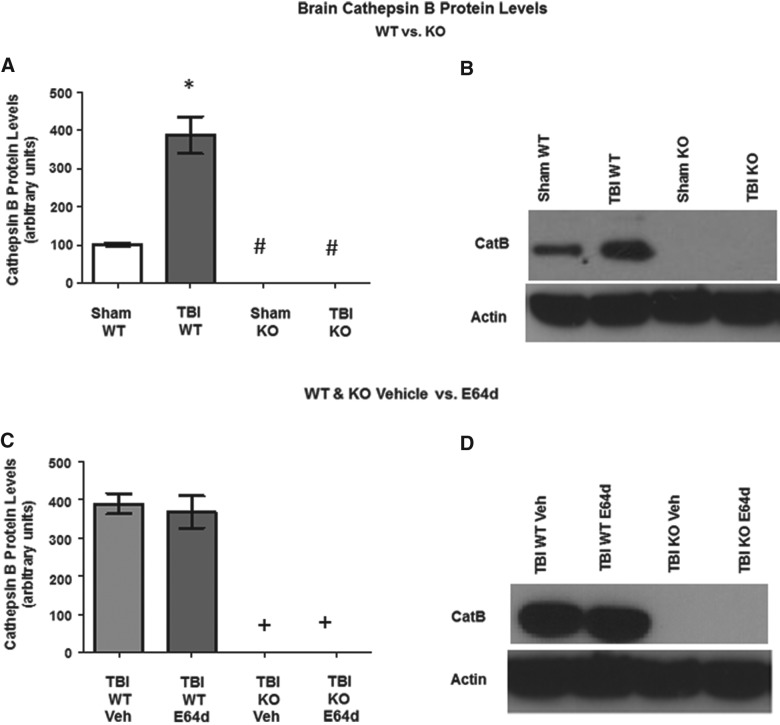
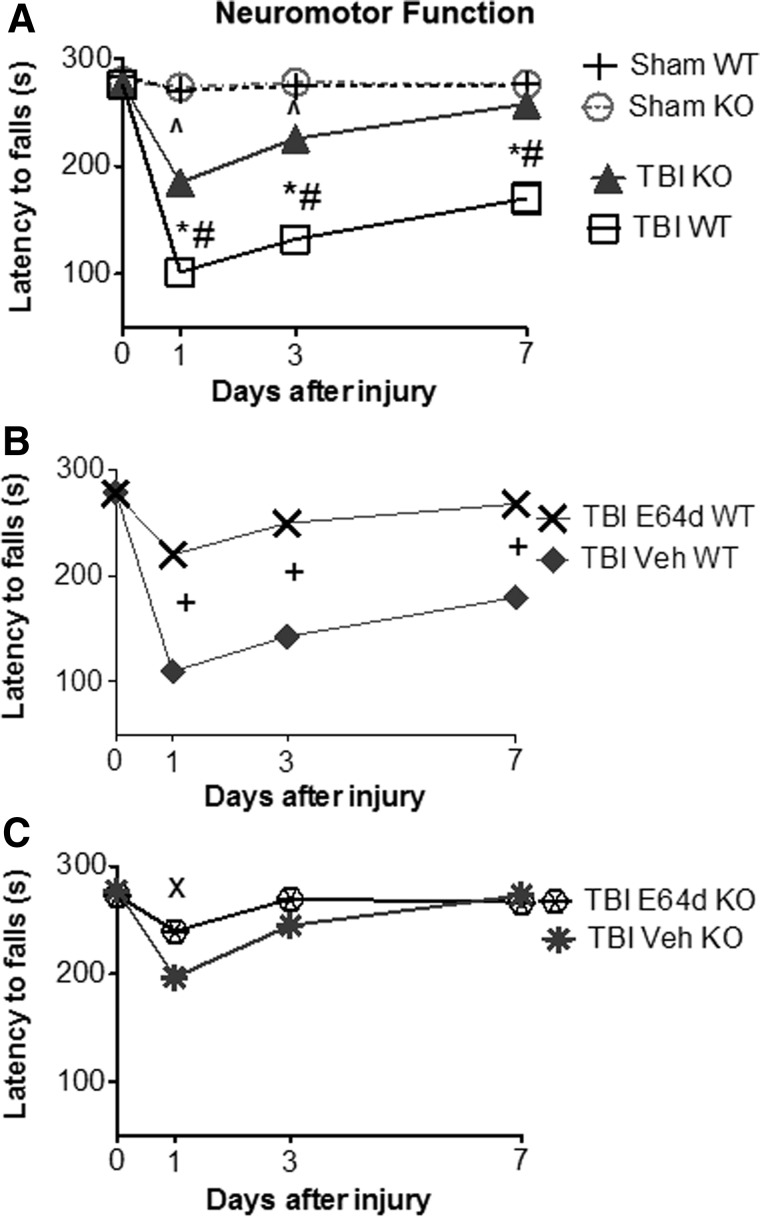
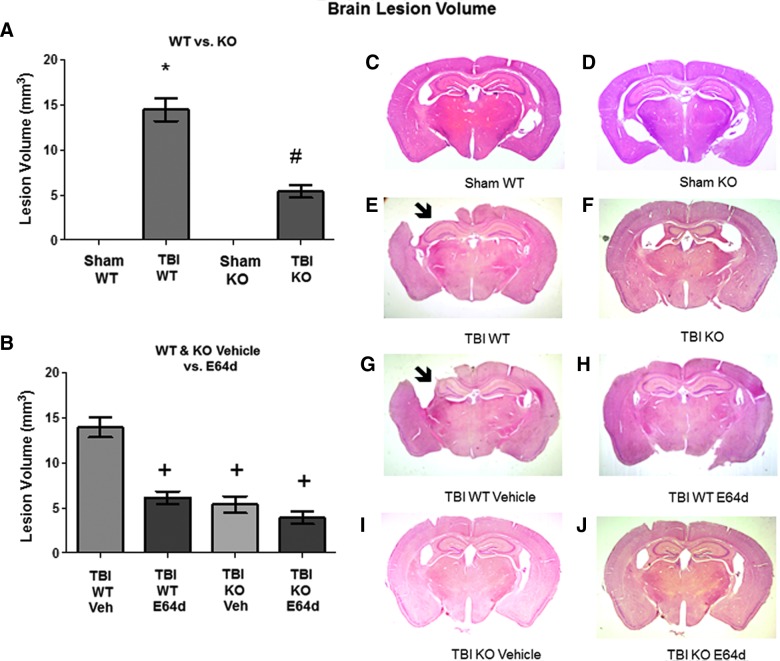
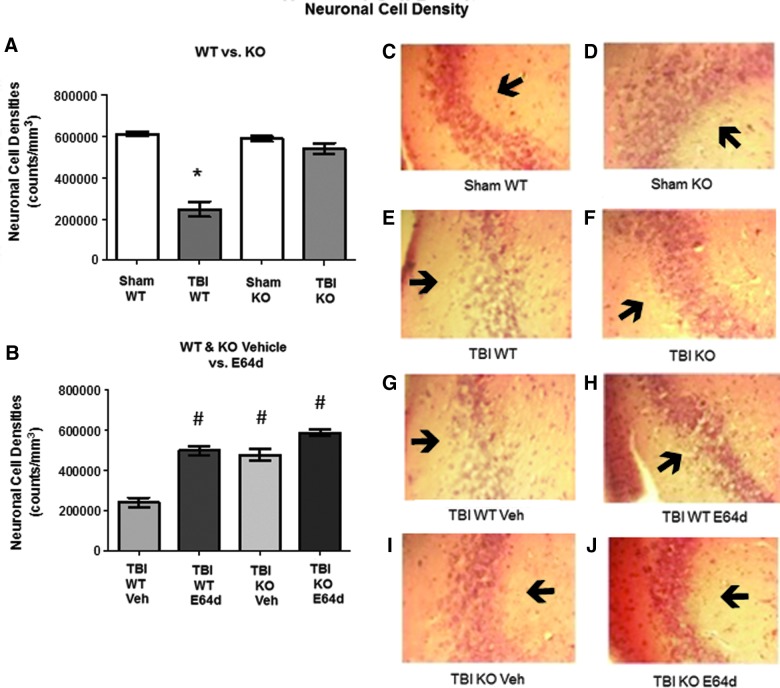
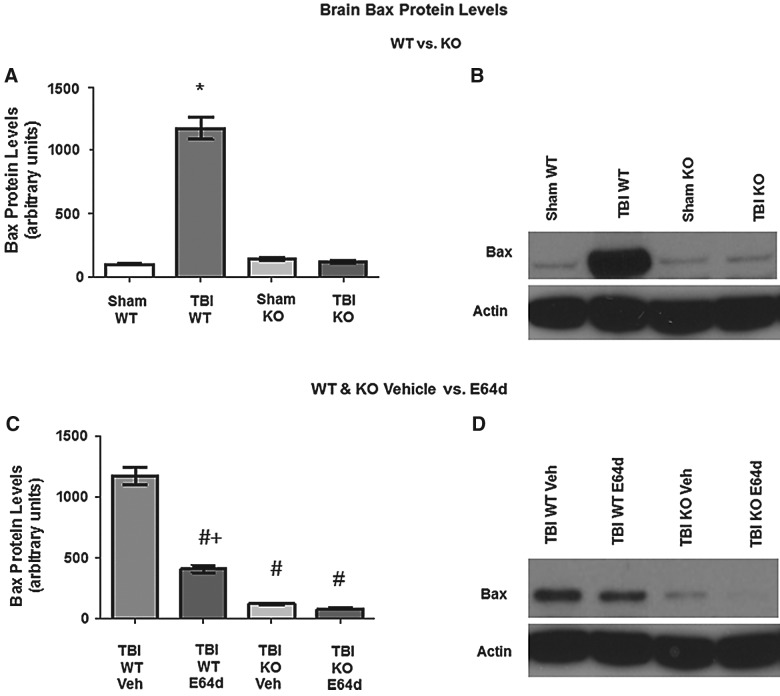
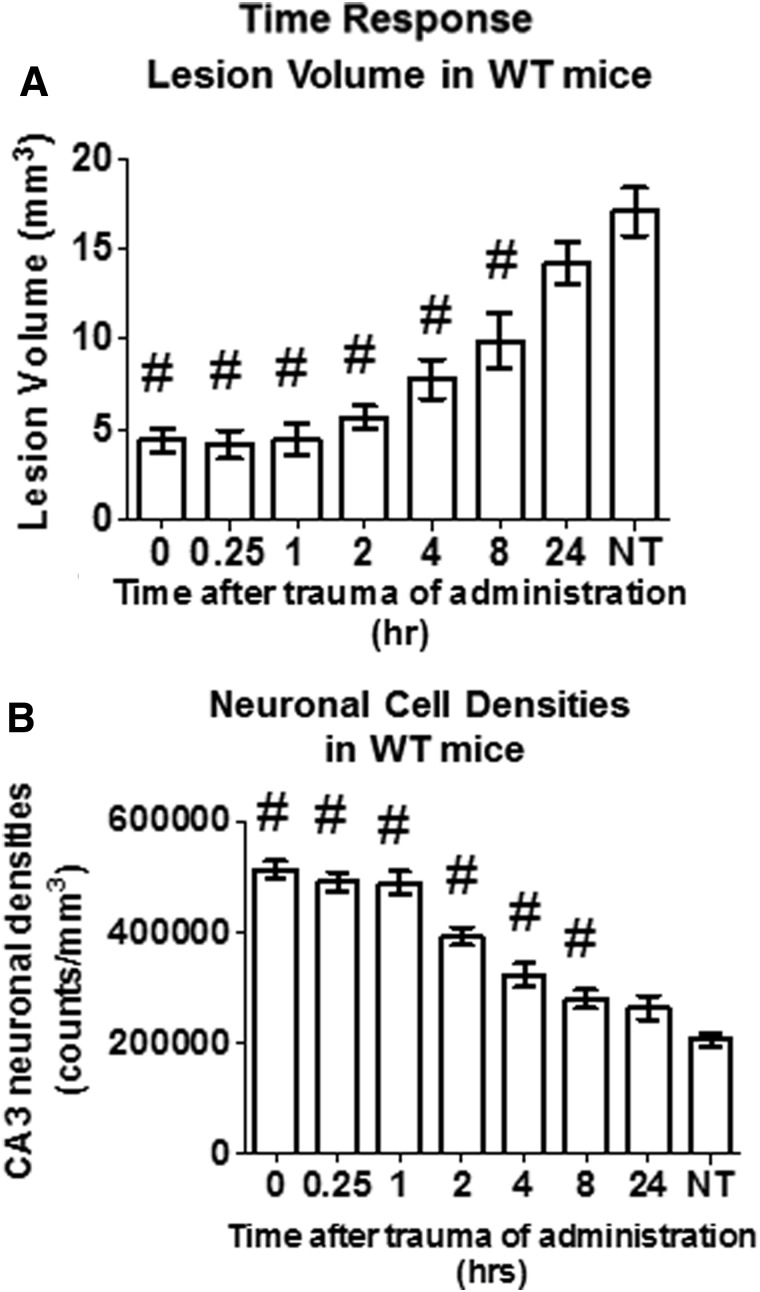
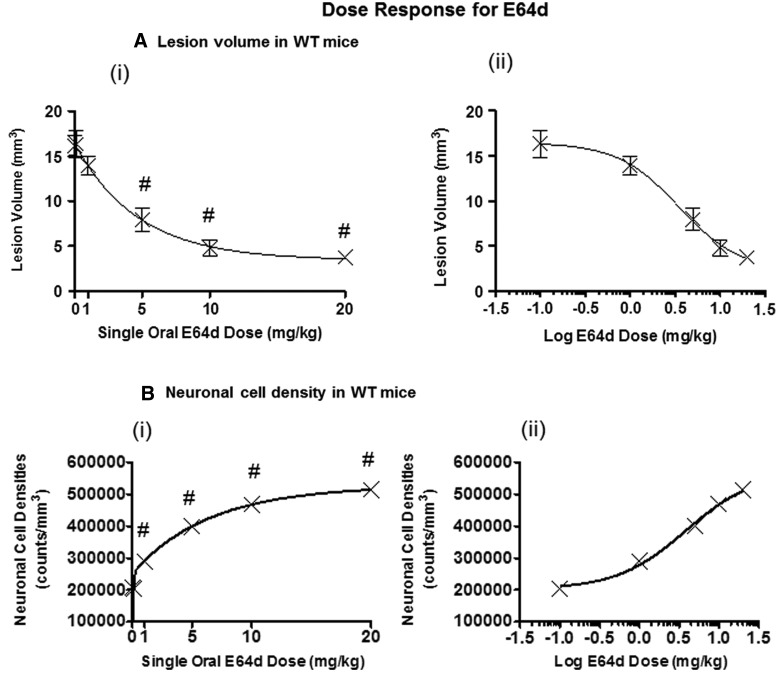
There are currently no effective therapeutic agents for traumatic brain injury (TBI), but drug treatments for TBI can be developed by validation of new drug targets and demonstration that compounds directed to such targets are efficacious in TBI animal models using a clinically relevant route of drug administration. The cysteine protease, cathepsin B, has been implicated in mediating TBI, but it has not been validated by gene knockout (KO) studies. Therefore, this investigation evaluated mice with deletion of the cathepsin B gene receiving controlled cortical impact TBI trauma. Results indicated that KO of the cathepsin B gene resulted in amelioration of TBI, shown by significant improvement in motor dysfunction, reduced brain lesion volume, greater neuronal density in brain, and lack of increased proapoptotic Bax levels. Notably, oral administration of the small-molecule cysteine protease inhibitor, E64d, immediately after TBI resulted in recovery of TBI-mediated motor dysfunction and reduced the increase in cathepsin B activity induced by TBI. E64d outcomes were as effective as cathepsin B gene deletion for improving TBI. E64d treatment was effective even when administered 8 h after injury, indicating a clinically plausible time period for acute therapeutic intervention. These data demonstrate that a cysteine protease inhibitor can be orally efficacious in a TBI animal model when administered at a clinically relevant time point post-trauma, and that E64d-mediated improvement of TBI is primarily the result of inhibition of cathepsin B activity. These results validate cathepsin B as a new TBI therapeutic target.
Figures
References
-
- Menon D.K., Schwab K., Wright D.W., and Maas A.I. (2010). Position statement: definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 91, 1637–1640 - PubMed
-
- Faul M., Xu L.X., Wald M., and Coronado V. (2010). Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Centers for Disease Control and Prevention, National Center for Intjury Prevention and Control: Atlanta, GA
-
- Rutland-Brown W., Langlois J.A., Thomas K.E., and Xi Y.L. (2006). Incidence of traumatic brain injury in the United States, 2003. J. Head Trauma Rehabil. 21, 544–548 - PubMed
-
- Warden D. (2006). Military TBI during the Iraq and Afghanistan wars. J. Head Trauma Rehabil. 21, 398–402 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
