

Exome sequencing reveals a thrombopoietin ligand mutation in a Micronesian family with autosomal recessive aplastic anemia
- PMID: 24085763
- PMCID: PMC3829117
- DOI: 10.1182/blood-2012-12-473538
Exome sequencing reveals a thrombopoietin ligand mutation in a Micronesian family with autosomal recessive aplastic anemia
Abstract
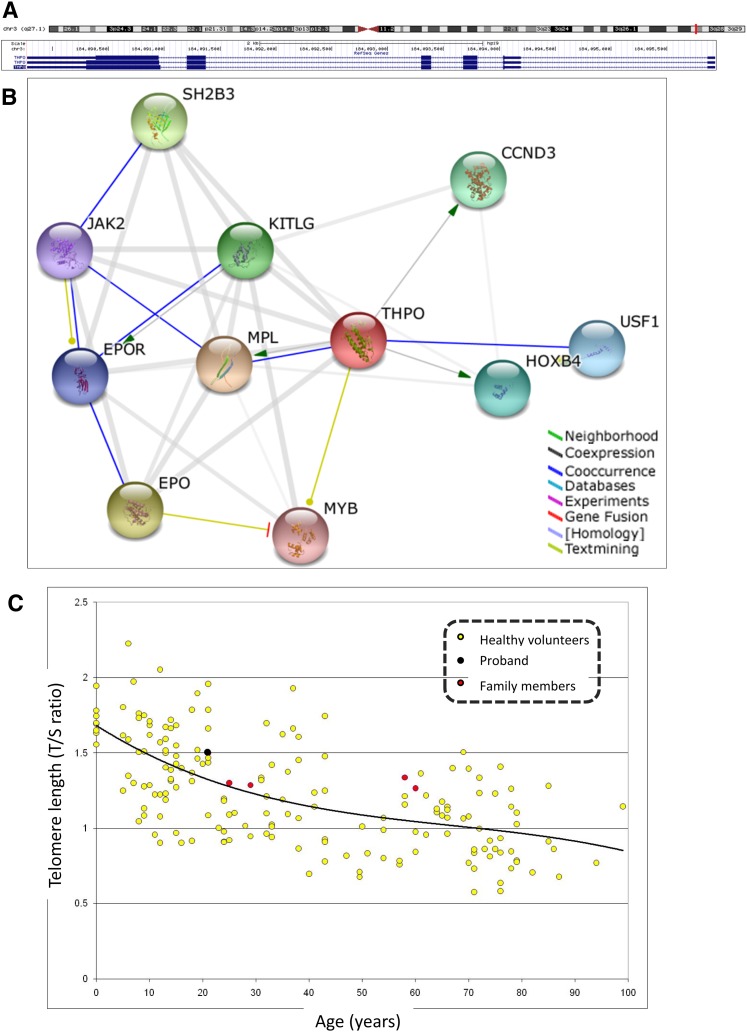
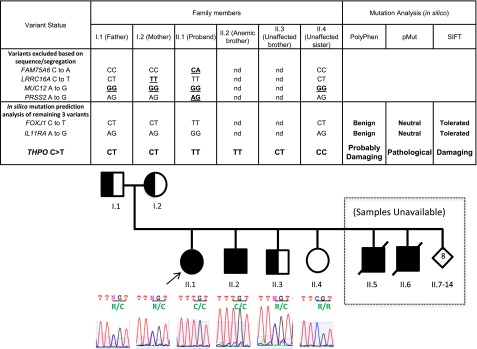
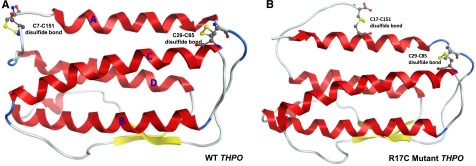
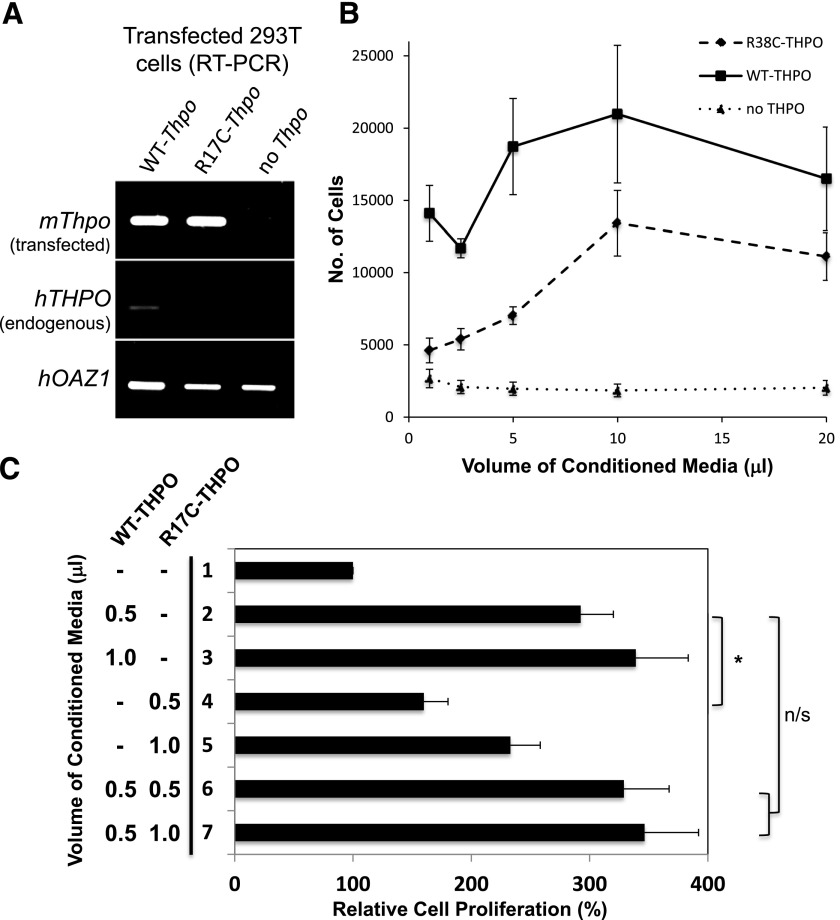
We recently identified 2 siblings afflicted with idiopathic, autosomal recessive aplastic anemia. Whole-exome sequencing identified a novel homozygous missense mutation in thrombopoietin (THPO, c.112C>T) in both affected siblings. This mutation encodes an arginine to cysteine substitution at residue 38 or residue 17 excluding the 21-amino acid signal peptide of THPO receptor binding domain (RBD). THPO has 4 conserved cysteines in its RBD that form 2 disulfide bonds. Our in silico modeling predicts that introduction of a fifth cysteine may disrupt normal disulfide bonding to cause poor receptor binding. In functional assays, the mutant-THPO-containing media shows two- to threefold reduced ability to sustain UT7-TPO cells, which require THPO for proliferation. Both parents and a sibling with heterozygous R17C change have reduced platelet counts, whereas a sibling with wild-type sequence has normal platelet count. Thus, the R17C partial loss-of-function allele results in aplastic anemia in the homozygous state and mild thrombocytopenia in the heterozygous state in our family. Together with the recent identification of THPO receptor (MPL) mutations and the effects of THPO agonists in aplastic anemia, our results have clinical implications in the diagnosis and treatment of patients with aplastic anemia and highlight a role for the THPO-MPL pathway in hematopoiesis in vivo.
Figures

References
-
- Calado RT. Telomeres and marrow failure. Hematology (Am Soc Hematol Educ Program) 2009:338–343. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
