

Nucleotide excision repair in eukaryotes
- PMID: 24086042
- PMCID: PMC3783044
- DOI: 10.1101/cshperspect.a012609
Nucleotide excision repair in eukaryotes
Abstract
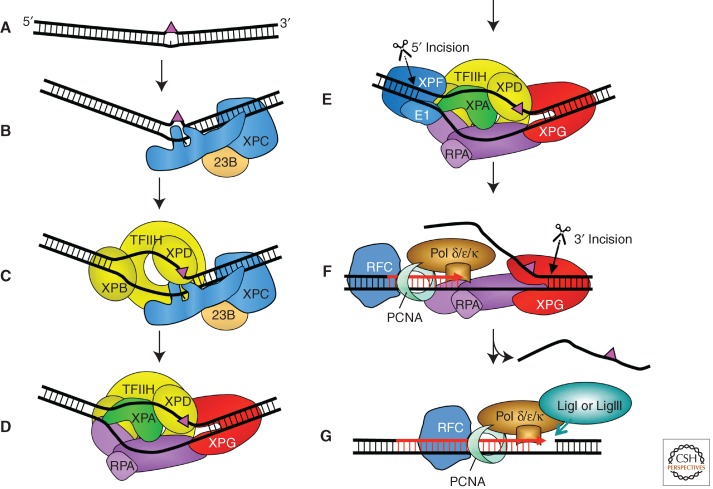
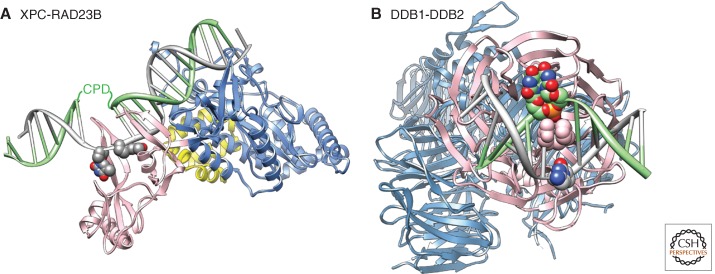
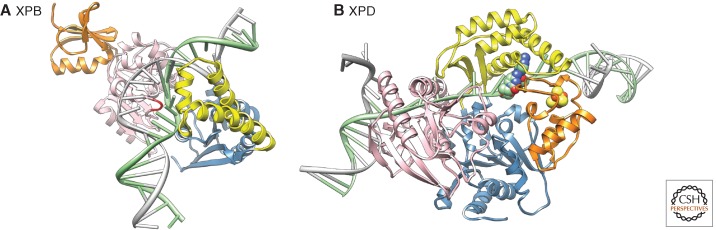
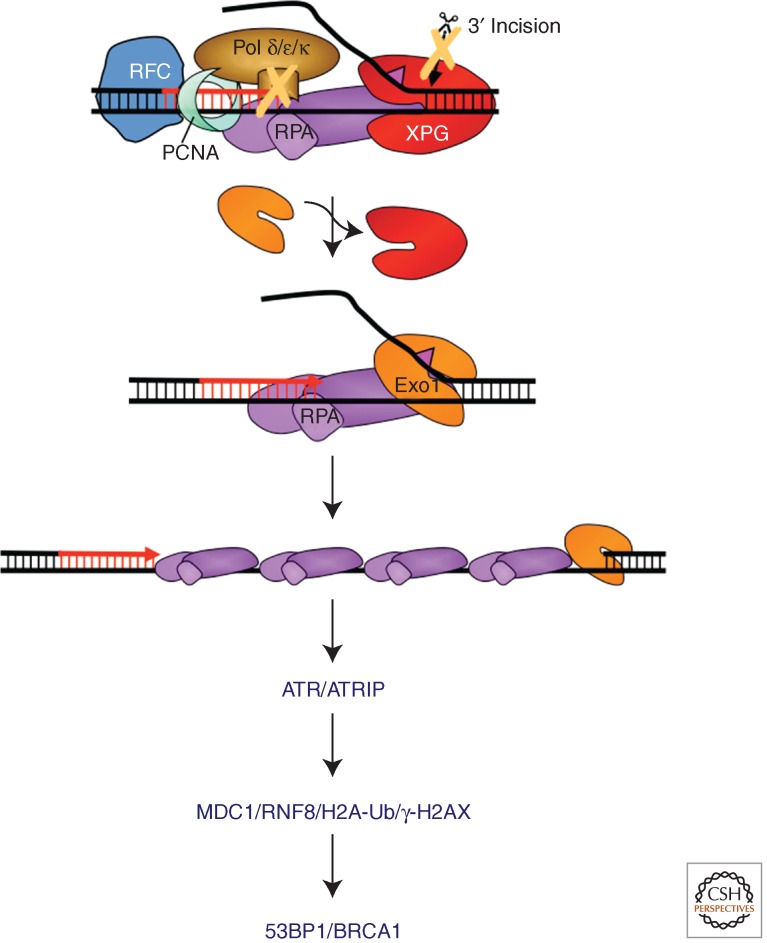
Nucleotide excision repair (NER) is the main pathway used by mammals to remove bulky DNA lesions such as those formed by UV light, environmental mutagens, and some cancer chemotherapeutic adducts from DNA. Deficiencies in NER are associated with the extremely skin cancer-prone inherited disorder xeroderma pigmentosum. Although the core NER reaction and the factors that execute it have been known for some years, recent studies have led to a much more detailed understanding of the NER mechanism, how NER operates in the context of chromatin, and how it is connected to other cellular processes such as DNA damage signaling and transcription. This review emphasizes biochemical, structural, cell biological, and genetic studies since 2005 that have shed light on many aspects of the NER pathway.
Figures
References
-
- Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD 1995. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 80: 859–868 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources