

Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein
- PMID: 24086135
- PMCID: PMC3784465
- DOI: 10.1371/journal.ppat.1003643
Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein
Abstract
Prions are infectious agents causing fatal neurodegenerative diseases of humans and animals. In humans, these have sporadic, acquired and inherited aetiologies. The inherited prion diseases are caused by one of over 30 coding mutations in the human prion protein (PrP) gene (PRNP) and many of these generate infectious prions as evidenced by their experimental transmissibility by inoculation to laboratory animals. However, some, and in particular an extensively studied type of Gerstmann-Sträussler-Scheinker syndrome (GSS) caused by a PRNP A117V mutation, are thought not to generate infectious prions and instead constitute prion proteinopathies with a quite distinct pathogenetic mechanism. Multiple attempts to transmit A117V GSS have been unsuccessful and typical protease-resistant PrP (PrP(Sc)), pathognomonic of prion disease, is not detected in brain. Pathogenesis is instead attributed to production of an aberrant topological form of PrP, C-terminal transmembrane PrP ((Ctm)PrP). Barriers to transmission of prion strains from one species to another appear to relate to structural compatibility of PrP in host and inoculum and we have therefore produced transgenic mice expressing human 117V PrP. We found that brain tissue from GSS A117V patients did transmit disease to these mice and both the neuropathological features of prion disease and presence of PrP(Sc) was demonstrated in the brains of recipient transgenic mice. This PrP(Sc) rapidly degraded during laboratory analysis, suggesting that the difficulty in its detection in patients with GSS A117V could relate to post-mortem proteolysis. We conclude that GSS A117V is indeed a prion disease although the relative contributions of (Ctm)PrP and prion propagation in neurodegeneration and their pathogenetic interaction remains to be established.
Conflict of interest statement
JC and JW are shareholders and JC is a director of D-Gen Limited, an academic spin-out company working in the field of prion disease diagnosis, decontamination and therapeutics. D-Gen markets one of the routine antibodies (ICSM 35) used in this study. This does not alter our adherence to all PLoS Pathogens policies on sharing data and materials.
Figures
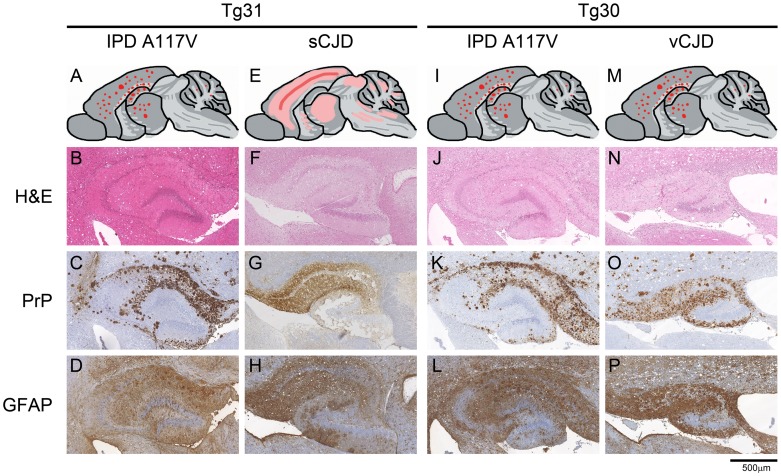
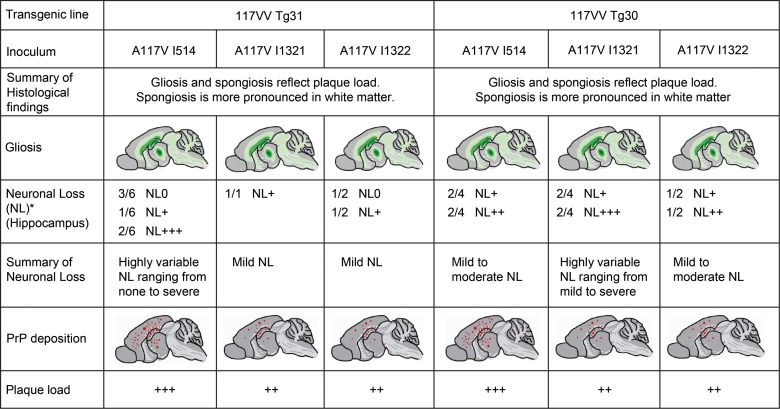
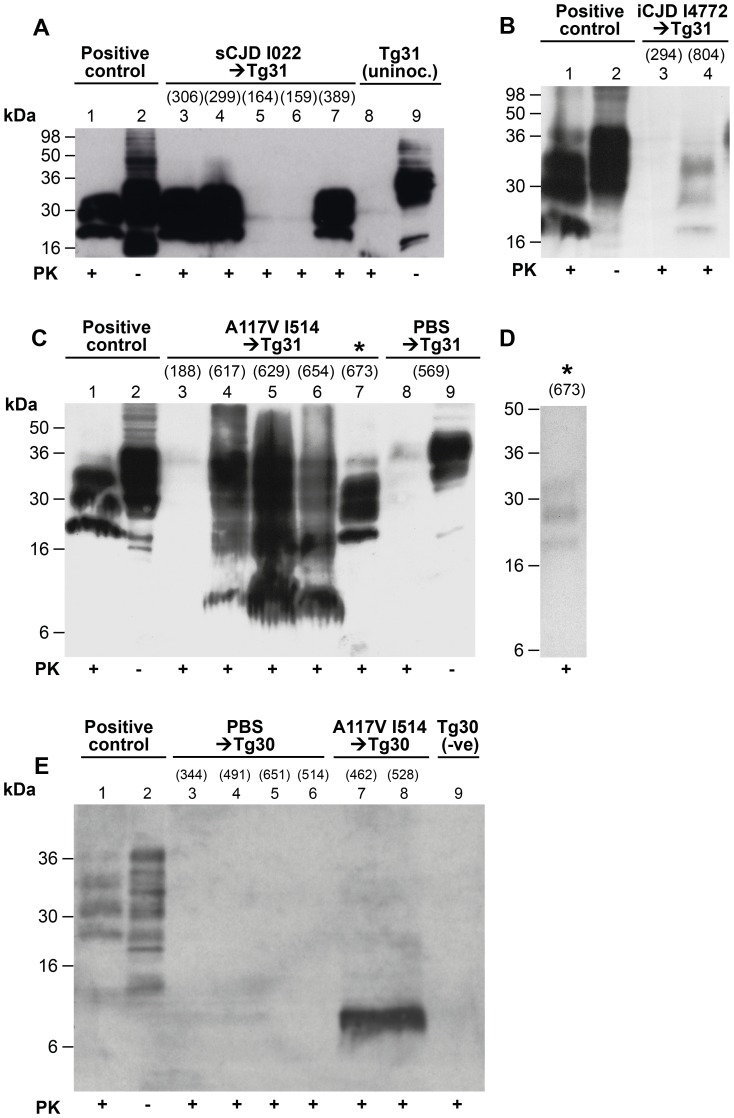

References
-
- Griffith JS (1967) Self replication and scrapie. Nature 215: 1043–1044. - PubMed
-
- Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216: 136–144. - PubMed
-
- Collinge J, Clarke A (2007) A general model of prion strains and their pathogenicity. Science 318: 930–936. - PubMed
-
- Collinge J (2001) Prion diseases of humans and animals: their causes and molecular basis. Ann Rev Neurosci 24: 519–550. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
