

microRNA-22 promotes heart failure through coordinate suppression of PPAR/ERR-nuclear hormone receptor transcription
- PMID: 24086656
- PMCID: PMC3785418
- DOI: 10.1371/journal.pone.0075882
microRNA-22 promotes heart failure through coordinate suppression of PPAR/ERR-nuclear hormone receptor transcription
Abstract
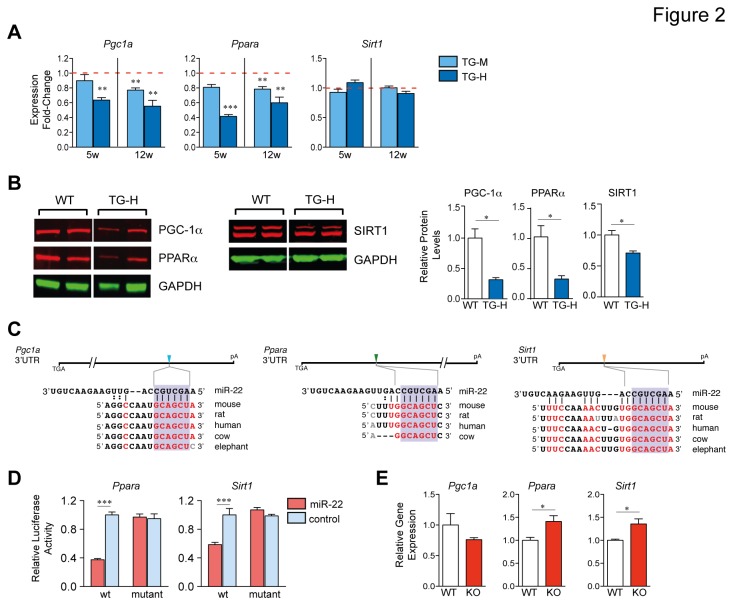
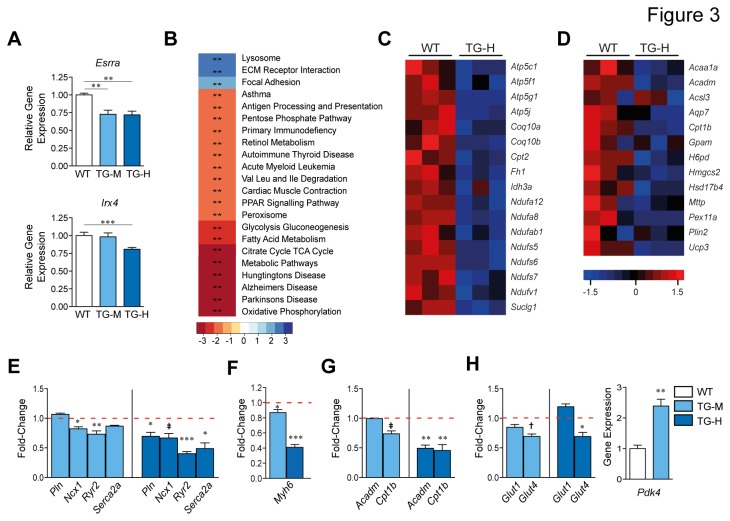
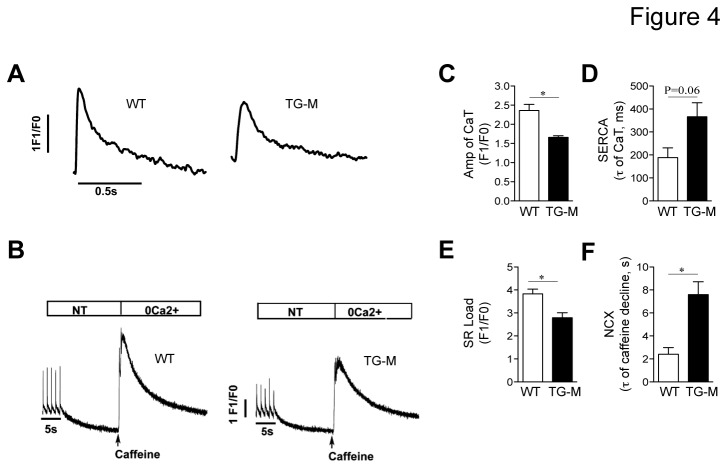
Increasing evidence suggests that microRNAs are intimately involved in the pathophysiology of heart failure. MicroRNA-22 (miR-22) is a muscle-enriched miRNA required for optimum cardiac gene transcription and adaptation to hemodynamic stress by pressure overload in mice. Recent evidence also suggests that miR-22 induces hypertrophic growth and it is oftentimes upregulated in end stage heart failure. However the scope of mRNA targets and networks of miR-22 in the heart failure remained unclear. We analyzed transgenic mice with enhanced levels of miR-22 expression in adult cardiomyocytes to identify important pathophysiologic targets of miR-22. Our data shows that forced expression of miR-22 induces a pro-hypertrophic gene expression program, and it elicits contractile dysfunction leading to cardiac dilation and heart failure. Increased expression of miR-22 impairs the Ca(2+) transient, Ca(2+) loading into the sarcoplasmic reticulum plus it interferes with transcription of estrogen related receptor (ERR) and PPAR downstream genes. Mechanistically, miR-22 postranscriptionally inhibits peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), PPARα and sirtuin 1 (SIRT1) expression via a synergistic circuit, which may account for deleterious actions of unchecked miR-22 expression on the heart.
Conflict of interest statement
Figures

References
-
- Diwan A, Dorn GW 2nd (2007) Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiol (Bethesda) 22: 56-64. doi:10.1152/physiol.00033.2006. PubMed: 17289931. - DOI - PubMed
-
- Frey N, Olson EN (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65: 45-79. doi:10.1146/annurev.physiol.65.092101.142243. PubMed: 12524460. - DOI - PubMed
-
- Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H (2007) Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev 12: 331-343. doi:10.1007/s10741-007-9034-1. PubMed: 17516164. - DOI - PubMed
-
- Heineke J, Molkentin JD (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7: 589-600. doi:10.1038/nrm1983. PubMed: 16936699. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL091947/HL/NHLBI NIH HHS/United States
- HL089598/HL/NHLBI NIH HHS/United States
- R01 HL22512/HL/NHLBI NIH HHS/United States
- K25 HL073041/HL/NHLBI NIH HHS/United States
- R01 HL022512/HL/NHLBI NIH HHS/United States
- R01 HL089598/HL/NHLBI NIH HHS/United States
- P41 RR011795/RR/NCRR NIH HHS/United States
- K25 HL73041/HL/NHLBI NIH HHS/United States
- P41 EB002182/EB/NIBIB NIH HHS/United States
- R01 HL089792/HL/NHLBI NIH HHS/United States
- R01 HL091947/HL/NHLBI NIH HHS/United States
- HL089792/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
