

Calculation of Tajima's D and other neutrality test statistics from low depth next-generation sequencing data
- PMID: 24088262
- PMCID: PMC4015034
- DOI: 10.1186/1471-2105-14-289
Calculation of Tajima's D and other neutrality test statistics from low depth next-generation sequencing data
Abstract
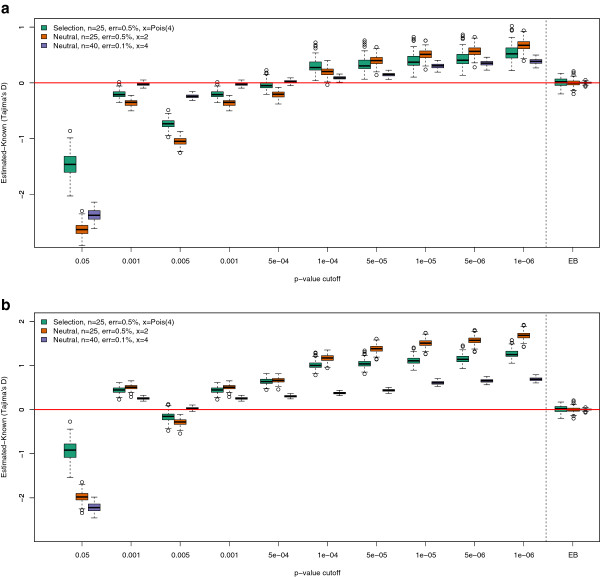
Background: A number of different statistics are used for detecting natural selection using DNA sequencing data, including statistics that are summaries of the frequency spectrum, such as Tajima's D. These statistics are now often being applied in the analysis of Next Generation Sequencing (NGS) data. However, estimates of frequency spectra from NGS data are strongly affected by low sequencing coverage; the inherent technology dependent variation in sequencing depth causes systematic differences in the value of the statistic among genomic regions.
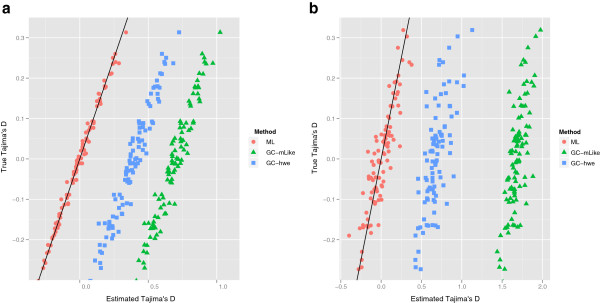
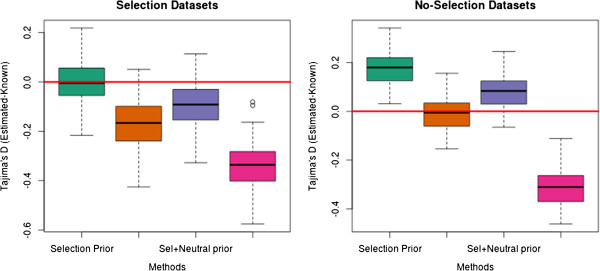
Results: We have developed an approach that accommodates the uncertainty of the data when calculating site frequency based neutrality test statistics. A salient feature of this approach is that it implicitly solves the problems of varying sequencing depth, missing data and avoids the need to infer variable sites for the analysis and thereby avoids ascertainment problems introduced by a SNP discovery process.
Conclusion: Using an empirical Bayes approach for fast computations, we show that this method produces results for low-coverage NGS data comparable to those achieved when the genotypes are known without uncertainty. We also validate the method in an analysis of data from the 1000 genomes project. The method is implemented in a fast framework which enables researchers to perform these neutrality tests on a genome-wide scale.
Figures





References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
