

The dynamics of genome replication using deep sequencing
- PMID: 24089142
- PMCID: PMC3874191
- DOI: 10.1093/nar/gkt878
The dynamics of genome replication using deep sequencing
Abstract
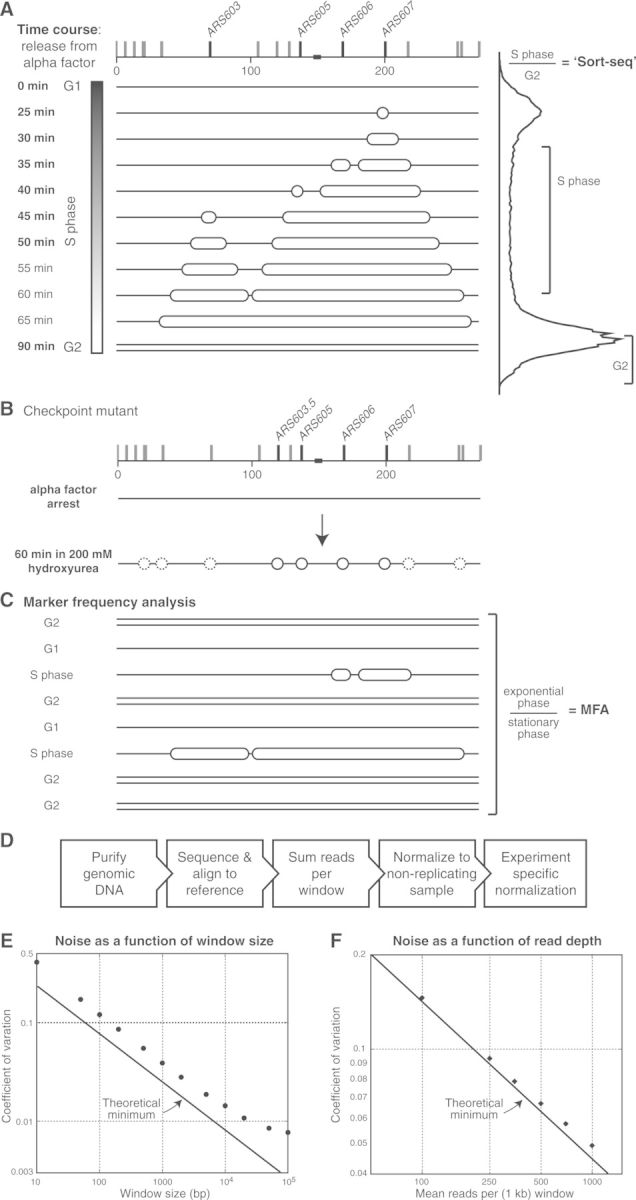
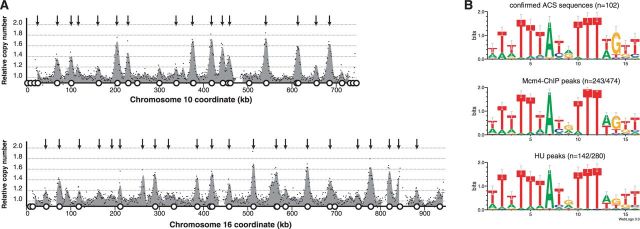
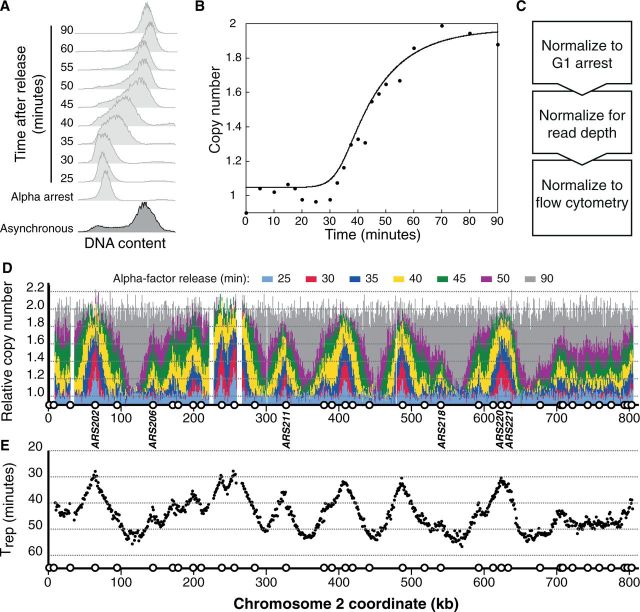
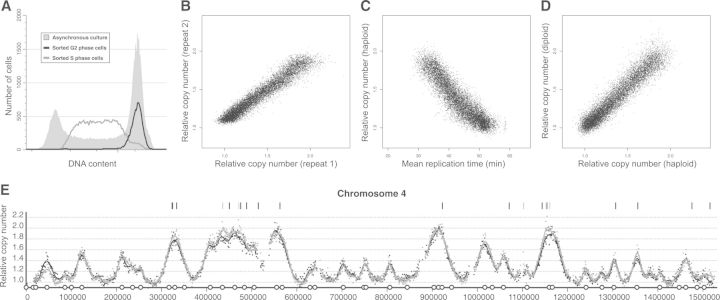
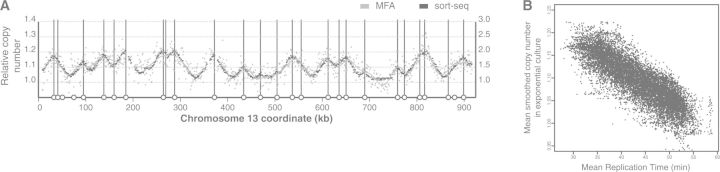
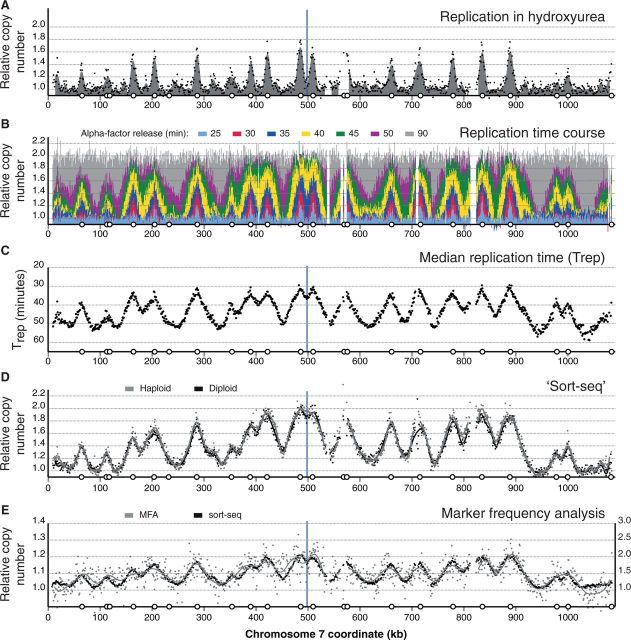
Eukaryotic genomes are replicated from multiple DNA replication origins. We present complementary deep sequencing approaches to measure origin location and activity in Saccharomyces cerevisiae. Measuring the increase in DNA copy number during a synchronous S-phase allowed the precise determination of genome replication. To map origin locations, replication forks were stalled close to their initiation sites; therefore, copy number enrichment was limited to origins. Replication timing profiles were generated from asynchronous cultures using fluorescence-activated cell sorting. Applying this technique we show that the replication profiles of haploid and diploid cells are indistinguishable, indicating that both cell types use the same cohort of origins with the same activities. Finally, increasing sequencing depth allowed the direct measure of replication dynamics from an exponentially growing culture. This is the first time this approach, called marker frequency analysis, has been successfully applied to a eukaryote. These data provide a high-resolution resource and methodological framework for studying genome biology.
Figures
References
-
- Mechali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010;11:728–738. - PubMed
-
- Mechali M, Yoshida K, Coulombe P, Pasero P. Genetic and epigenetic determinants of DNA replication origins, position and activation. Curr. Opin. Genet. Dev. 2013;23:124–131. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
Grants and funding
- BB/K007211/2/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/G001596/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- G0900747/MRC_/Medical Research Council/United Kingdom
- BB/E023754/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/F00513X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
