

Empirical validation of viral quasispecies assembly algorithms: state-of-the-art and challenges
- PMID: 24089188
- PMCID: PMC3789152
- DOI: 10.1038/srep02837
Empirical validation of viral quasispecies assembly algorithms: state-of-the-art and challenges
Abstract
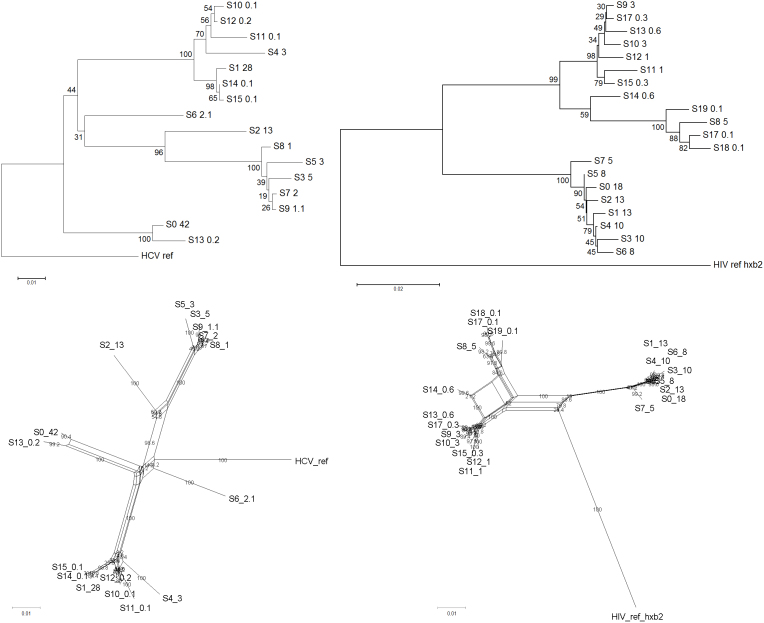
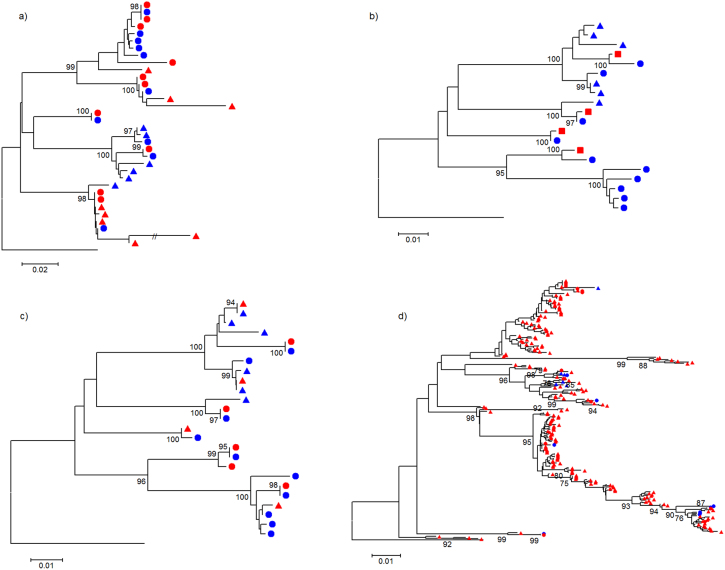
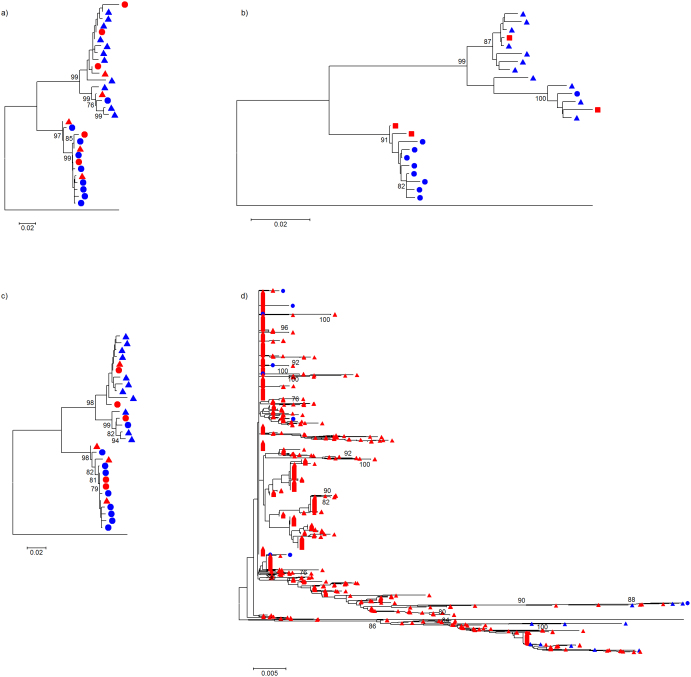
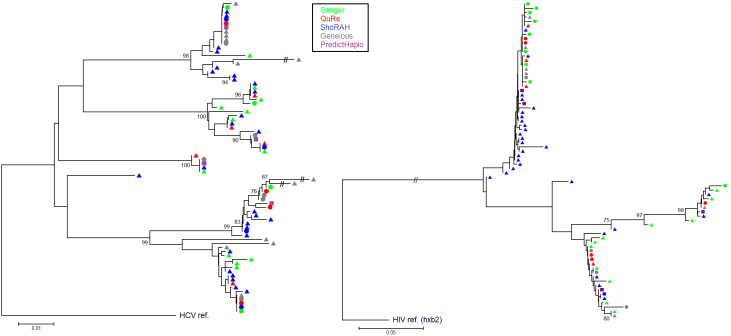
Next generation sequencing (NGS) is superseding Sanger technology for analysing intra-host viral populations, in terms of genome length and resolution. We introduce two new empirical validation data sets and test the available viral population assembly software. Two intra-host viral population 'quasispecies' samples (type-1 human immunodeficiency and hepatitis C virus) were Sanger-sequenced, and plasmid clone mixtures at controlled proportions were shotgun-sequenced using Roche's 454 sequencing platform. The performance of different assemblers was compared in terms of phylogenetic clustering and recombination with the Sanger clones. Phylogenetic clustering showed that all assemblers captured a proportion of the most divergent lineages, but none were able to provide a high precision/recall tradeoff. Estimated variant frequencies mildly correlated with the original. Given the limitations of currently available algorithms identified by our empirical validation, the development and exploitation of additional data sets is needed, in order to establish an efficient framework for viral population reconstruction using NGS.
Figures
Similar articles
-
Combinatorial analysis and algorithms for quasispecies reconstruction using next-generation sequencing.BMC Bioinformatics. 2011 Jan 5;12:5. doi: 10.1186/1471-2105-12-5. BMC Bioinformatics. 2011. PMID: 21208435 Free PMC article.
-
Streamlined Subpopulation, Subtype, and Recombination Analysis of HIV-1 Half-Genome Sequences Generated by High-Throughput Sequencing.mSphere. 2020 Oct 14;5(5):e00551-20. doi: 10.1128/mSphere.00551-20. mSphere. 2020. PMID: 33055255 Free PMC article.
-
An Oxford Nanopore Technology-Based Hepatitis B Virus Sequencing Protocol Suitable for Genomic Surveillance Within Clinical Diagnostic Settings.Int J Mol Sci. 2024 Oct 31;25(21):11702. doi: 10.3390/ijms252111702. Int J Mol Sci. 2024. PMID: 39519254 Free PMC article.
-
Applications of next-generation sequencing technologies to diagnostic virology.Int J Mol Sci. 2011;12(11):7861-84. doi: 10.3390/ijms12117861. Epub 2011 Nov 14. Int J Mol Sci. 2011. PMID: 22174638 Free PMC article. Review.
-
Within-host nucleotide diversity of virus populations: insights from next-generation sequencing.Infect Genet Evol. 2015 Mar;30:1-7. doi: 10.1016/j.meegid.2014.11.026. Epub 2014 Dec 4. Infect Genet Evol. 2015. PMID: 25481279 Free PMC article. Review.
Cited by
-
PIQMEE: Bayesian Phylodynamic Method for Analysis of Large Data Sets with Duplicate Sequences.Mol Biol Evol. 2020 Oct 1;37(10):3061-3075. doi: 10.1093/molbev/msaa136. Mol Biol Evol. 2020. PMID: 32492139 Free PMC article.
-
Full-length haplotype reconstruction to infer the structure of heterogeneous virus populations.Nucleic Acids Res. 2014 Aug;42(14):e115. doi: 10.1093/nar/gku537. Epub 2014 Jun 27. Nucleic Acids Res. 2014. PMID: 24972832 Free PMC article.
-
De novo assembly of highly polymorphic metagenomic data using in situ generated reference sequences and a novel BLAST-based assembly pipeline.BMC Bioinformatics. 2017 Apr 26;18(1):223. doi: 10.1186/s12859-017-1630-z. BMC Bioinformatics. 2017. PMID: 28446139 Free PMC article.
-
Separation and assembly of deep sequencing data into discrete sub-population genomes.Nucleic Acids Res. 2017 Nov 2;45(19):10989-11003. doi: 10.1093/nar/gkx755. Nucleic Acids Res. 2017. PMID: 28977510 Free PMC article.
-
Towards Better Precision Medicine: PacBio Single-Molecule Long Reads Resolve the Interpretation of HIV Drug Resistant Mutation Profiles at Explicit Quasispecies (Haplotype) Level.J Data Mining Genomics Proteomics. 2016 Jan;7(1):182. doi: 10.4172/2153-0602.1000182. Epub 2015 Nov 8. J Data Mining Genomics Proteomics. 2016. PMID: 26949565 Free PMC article.
References
-
- Metzker M. L. Sequencing technologies - the next generation. Nat Rev Genet 11, 31–46 (2010). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
