

Herpes simplex virus 1 ICP22 but not US 1.5 is required for efficient acute replication in mice and VICE domain formation
- PMID: 24089574
- PMCID: PMC3838291
- DOI: 10.1128/JVI.02424-13
Herpes simplex virus 1 ICP22 but not US 1.5 is required for efficient acute replication in mice and VICE domain formation
Abstract
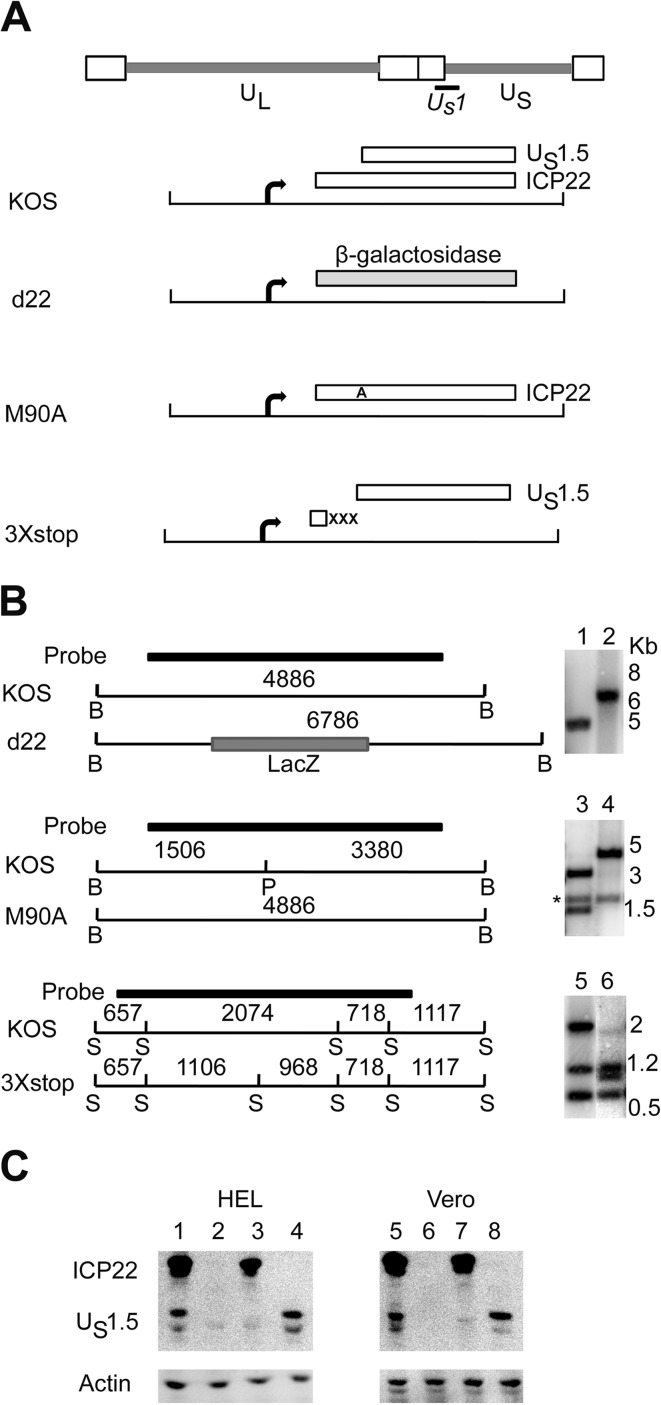
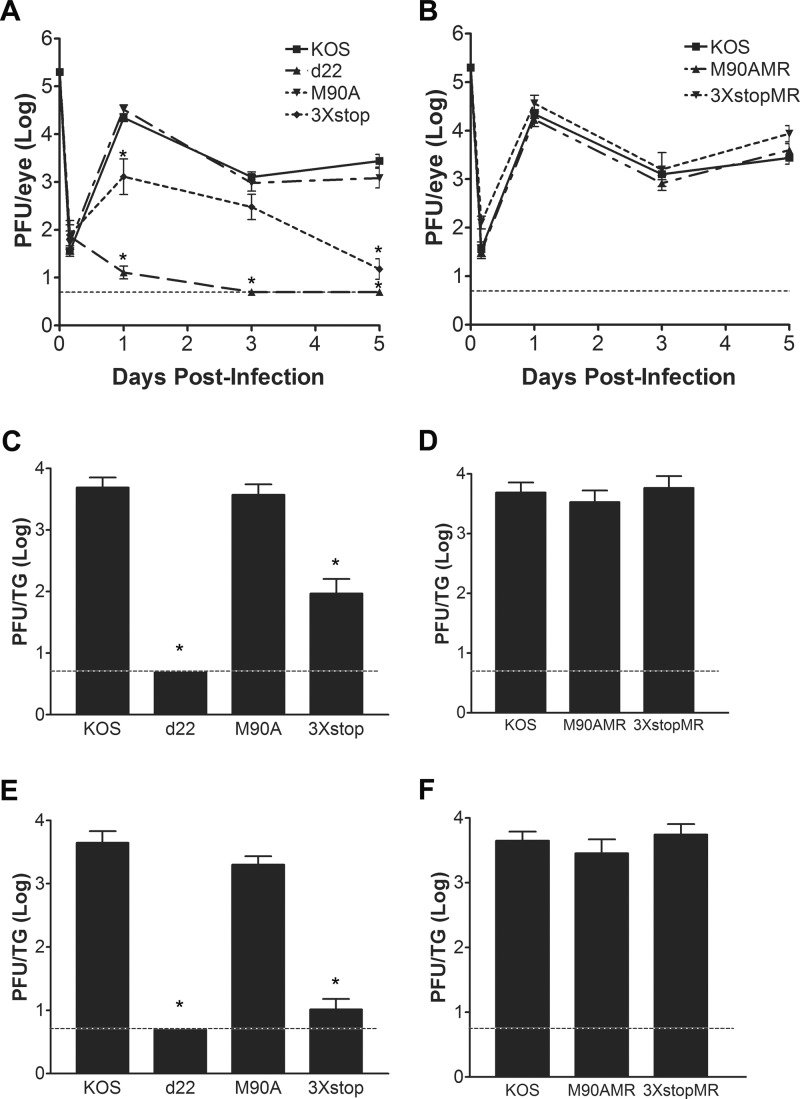
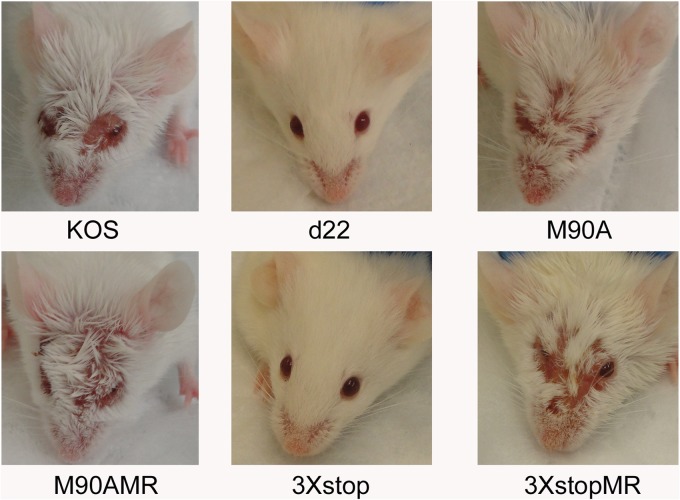
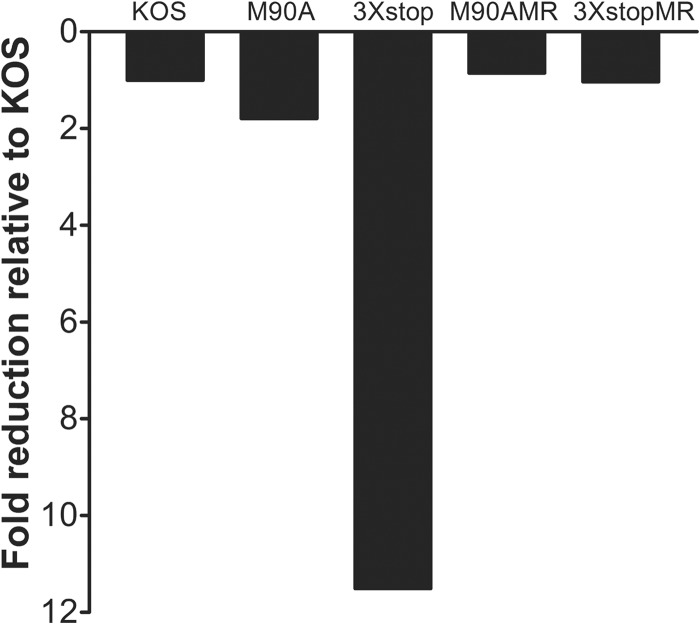
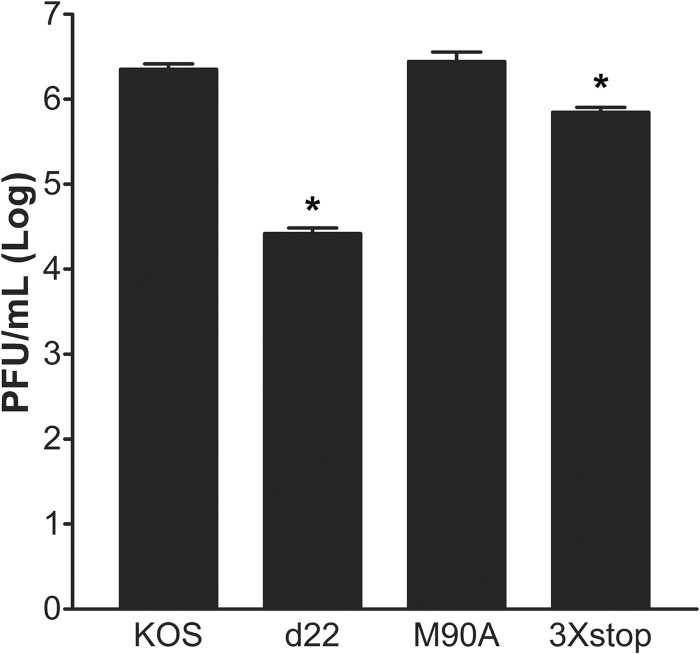
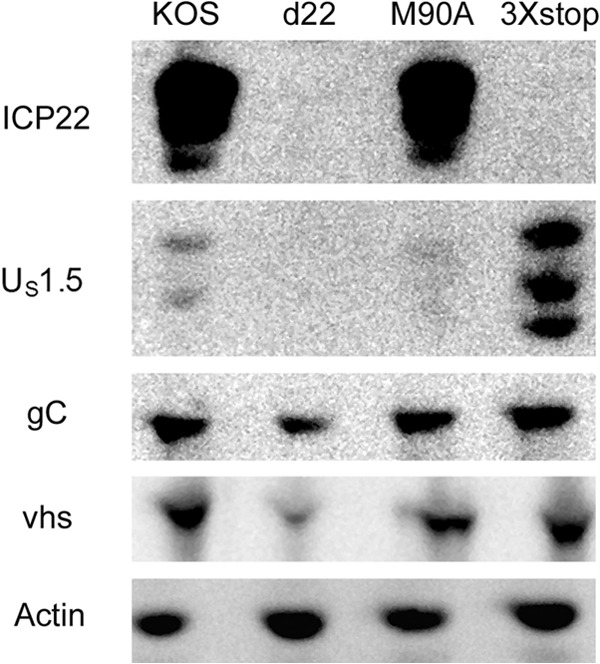
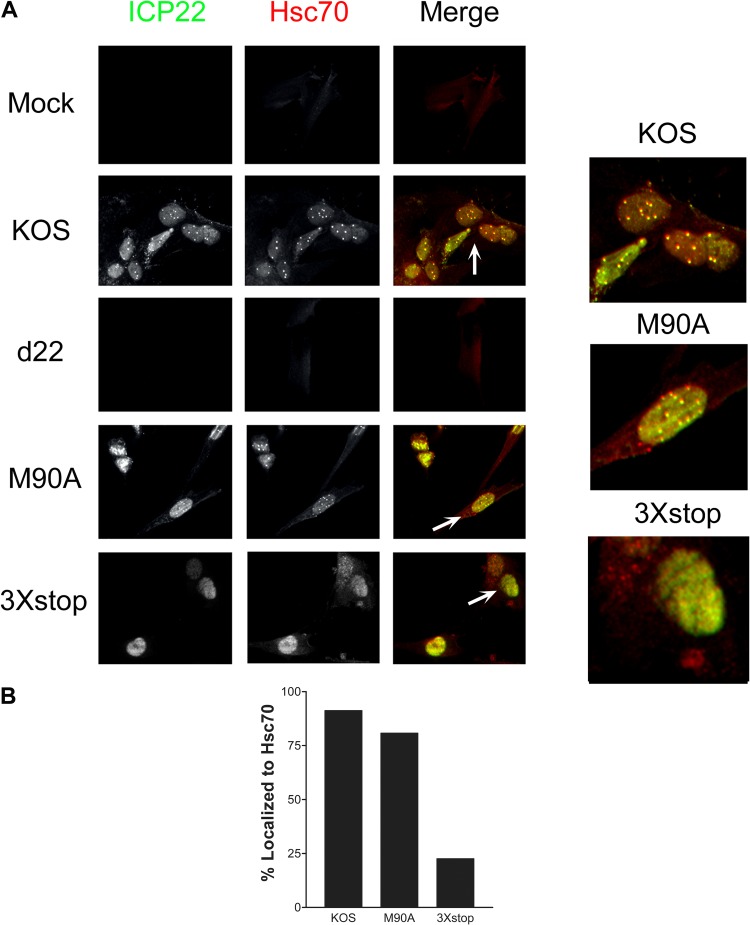
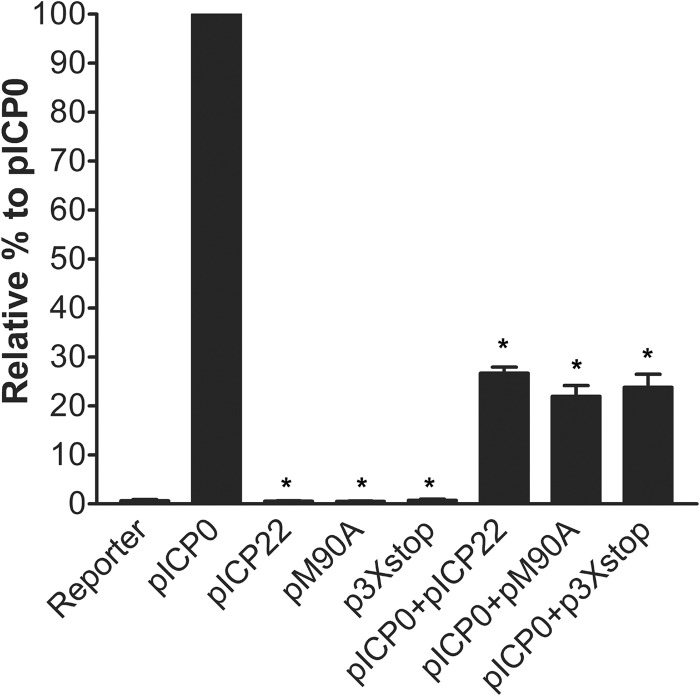
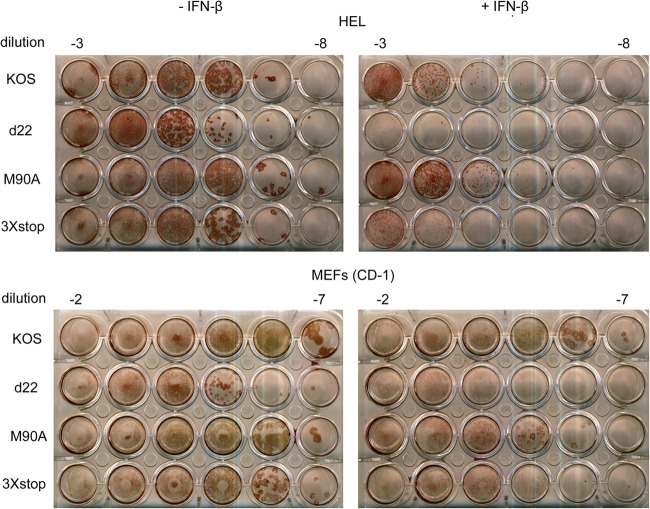
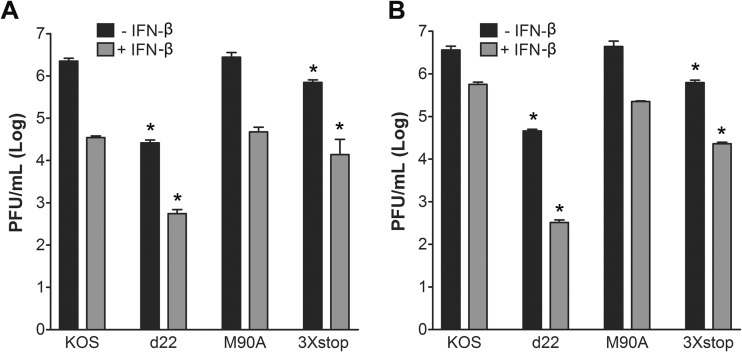
The herpes simplex virus 1 (HSV-1) immediate-early protein, infected cell protein 22 (ICP22), is required for efficient replication in restrictive cells, for virus-induced chaperone-enriched (VICE) domain formation, and for normal expression of a subset of viral late proteins. Additionally, ICP22 is important for optimal acute viral replication in vivo. Previous studies have shown that the US1 gene that encodes ICP22, produces an in-frame, N-terminally truncated form of ICP22, known as US1.5. To date, studies conducted to characterize the functions of ICP22 have not separated its functions from those of US1.5. To determine the individual roles of ICP22 and US1.5, we made viral mutants that express either ICP22 with an M90A mutation in the US1.5 initiation codon (M90A) or US1.5 with three stop codons introduced upstream of the US1.5 start codon (3×stop). Our studies showed that, in contrast to M90A, 3×stop was unable to replicate efficiently in the eyes and trigeminal ganglia of mice during acute infection, to efficiently establish a latent infection, or to induce VICE domain formation and was only mildly reduced in its replication in restrictive HEL-299 cells and murine embryonic fibroblasts (MEFs). Both mutants enhanced the expression of the late viral proteins virion host shutoff (vhs) and glycoprotein C (gC) and inhibited viral gene expression mediated by HSV-1 infected cell protein 0 (ICP0). When we tested our mutants' sensitivity to type I interferon (beta interferon [IFN-β]) in restrictive cells, we noticed that the plating of the ICP22 null (d22) and 3×stop mutants was reduced by the addition of IFN-β. Overall, our data suggest that US1.5 partially complements the functions of ICP22.
Figures
Similar articles
-
Functional anatomy of herpes simplex virus 1 overlapping genes encoding infected-cell protein 22 and US1.5 protein.J Virol. 1999 May;73(5):4305-15. doi: 10.1128/JVI.73.5.4305-4315.1999. J Virol. 1999. PMID: 10196329 Free PMC article.
-
Herpes simplex virus type 1 immediate-early protein ICP22 is required for VICE domain formation during productive viral infection.J Virol. 2010 Mar;84(5):2384-94. doi: 10.1128/JVI.01686-09. Epub 2009 Dec 23. J Virol. 2010. PMID: 20032172 Free PMC article.
-
The Herpes Simplex Virus 1 Immediate Early Protein ICP22 Is a Functional Mimic of a Cellular J Protein.J Virol. 2020 Jan 31;94(4):e01564-19. doi: 10.1128/JVI.01564-19. Print 2020 Jan 31. J Virol. 2020. PMID: 31748398 Free PMC article.
-
"Non-Essential" Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review.Viruses. 2020 Dec 23;13(1):17. doi: 10.3390/v13010017. Viruses. 2020. PMID: 33374862 Free PMC article. Review.
-
HSV-1 ICP22: hijacking host nuclear functions to enhance viral infection.Future Microbiol. 2013 Mar;8(3):311-21. doi: 10.2217/fmb.13.4. Future Microbiol. 2013. PMID: 23464370 Review.
Cited by
-
ICP22/IE63 Mediated Transcriptional Regulation and Immune Evasion: Two Important Survival Strategies for Alphaherpesviruses.Front Immunol. 2021 Dec 2;12:743466. doi: 10.3389/fimmu.2021.743466. eCollection 2021. Front Immunol. 2021. PMID: 34925320 Free PMC article. Review.
-
HSV-1 ICP22 condensates impair host transcription by depleting promoter RNAPII Ser-2P occupation.Front Microbiol. 2025 Feb 18;16:1538737. doi: 10.3389/fmicb.2025.1538737. eCollection 2025. Front Microbiol. 2025. PMID: 40041865 Free PMC article.
-
HSV-1 ICP22 Is a Selective Viral Repressor of Cellular RNA Polymerase II-Mediated Transcription Elongation.Vaccines (Basel). 2021 Sep 22;9(10):1054. doi: 10.3390/vaccines9101054. Vaccines (Basel). 2021. PMID: 34696162 Free PMC article.
-
Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location.Viruses. 2020 Jan 29;12(2):151. doi: 10.3390/v12020151. Viruses. 2020. PMID: 32013091 Free PMC article. Review.
-
P-TEFb goes viral.Inside Cell. 2016 Apr;1(2):106-116. doi: 10.1002/icl3.1037. Epub 2015 Nov 25. Inside Cell. 2016. PMID: 27398404 Free PMC article. Review.
References
-
- Roizman R, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA
-
- Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 6:211–221 - PubMed
-
- Roizman B, Kozak M, Honess RW, Hayward G. 1975. Regulation of herpesvirus macromolecular synthesis: evidence for multilevel regulation of herpes simplex 1 RNA and protein synthesis. Cold Spring Harbor Symp. Quant. Biol. 39:687–701 - PubMed
-
- Stevens JG. 1987. Defining herpes simplex genes involved in neurovirulence and neuroinvasiveness. Curr. Eye Res. 6:63–67 - PubMed
-
- Post LE, Roizman B. 1981. A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell 25:227–232 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
