

Particular Mal de Meleda phenotypes in Tunisia and mutations founder effect in the Mediterranean region
- PMID: 24093092
- PMCID: PMC3777190
- DOI: 10.1155/2013/206803
Particular Mal de Meleda phenotypes in Tunisia and mutations founder effect in the Mediterranean region
Abstract
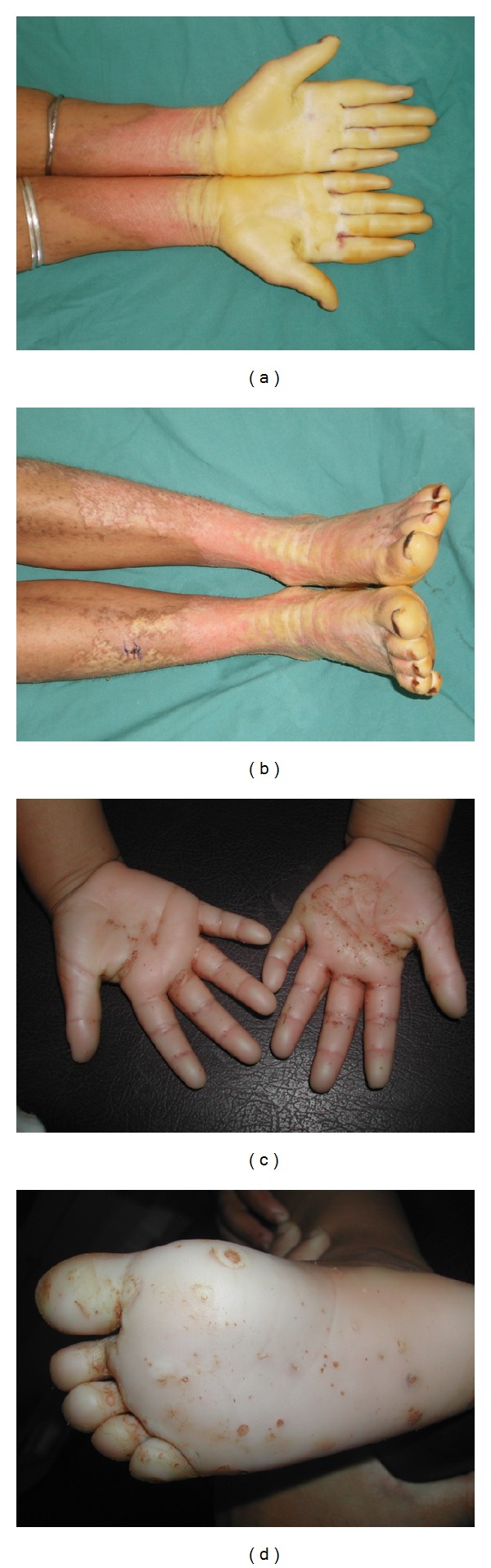
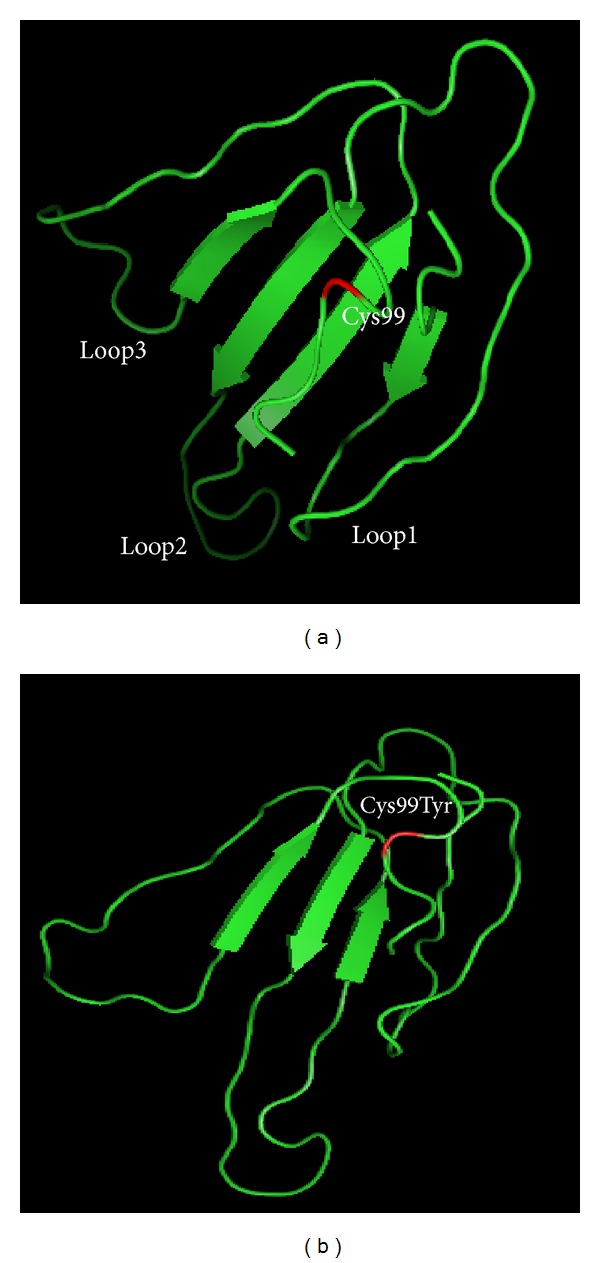
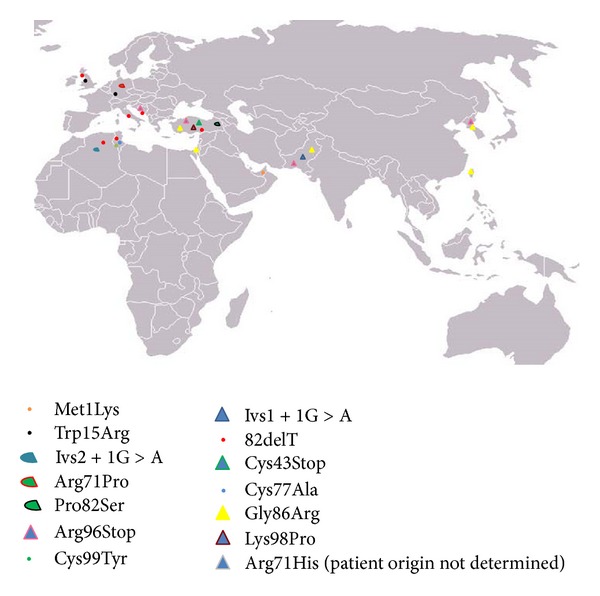
Mal de Meleda (MDM) is a rare, autosomal recessive form of palmoplantar keratoderma. It is characterized by erythema and hyperkeratosis of the palms and soles that progressively extend to the dorsal surface of the hands and feet. It is caused by mutations in SLURP-1 gene encoding for secreted mammalian Ly-6/uPAR-related protein 1 (SLURP-1). We performed mutational analysis by direct sequencing of SLURP-1 gene in order to identify the genetic defect in three unrelated families (families MDM-12, MDM-13, and MDM-14) variably affected with transgressive palmoplantar keratoderma. A spectrum of clinical presentations with variable features has been observed from the pronounced to the transparent hyperkeratosis. We identified the 82delT frame shift mutation in the SLURP-1 gene in both families MDM-12 and MDM-13 and the missense variation p.Cys99Tyr in family MDM-14. To date, the 82delT variation is the most frequent cause of MDM in the world which is in favour of a recurrent molecular defect. The p.Cys99Tyr variation is only described in Tunisian families making evidence of founder effect mutation of likely Tunisian origin. Our patients presented with very severe to relatively mild phenotypes, including multiple keratolytic pits observed for one patient in the hyperkeratotic area which was not previously reported. The phenotypic variability may reflect the influence of additional factors on disease characteristics. This report further expands the spectrum of clinical phenotypes associated with mutations in SLURP1 in the Mediterranean population.
Figures
References
-
- Romdhane L, Abdelhak S. Genetic Disorders in North African Populations. Oxford University Press; 2012. (Genomics and Health in the Developing World).
-
- Bouadjar B, Benmazouzia S, Prud’homme J-F, Cure S, Fischer J. Clinical and genetic studies of 3 large, consanguineous, Algerian families with Mal de Meleda. Archives of Dermatology. 2000;136(10):1247–1252. - PubMed
-
- Marrakchi S, Audebert S, Bouadjar B, et al. Novel mutations in the gene encoding secreted lymphocyte antigen-6/urokinase-type plasminogen activator receptor-related protein-1 (SLURP-1) and description of five ancestral haplotypes in patients with Mal de Meleda. Journal of Investigative Dermatology. 2003;120(3):351–355. - PubMed
-
- Charfeddine C, Mokni M, Ben Mousli R, et al. A novel missense mutation in the gene encoding SLURP-1 in patients with Mal de Meleda from northern Tunisia. British Journal of Dermatology. 2003;149(6):1108–1115. - PubMed
-
- Frenk E, Guggisberg D, Mevorah B, Hohl D. Meleda disease: report of two cases investigated by electron microscopy. Dermatology. 1996;193(4):358–361. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
