

The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads
- PMID: 24093210
- PMCID: PMC3852528
- DOI: 10.1186/1471-2164-14-686
The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads
Abstract
Background: Next generation sequencing provides a powerful tool to study genome structure in species whose genomes are far from being completely sequenced. In this work we describe and compare different computational approaches to evaluate the repetitive component of the genome of sunflower, by using medium/low coverage Illumina or 454 libraries.
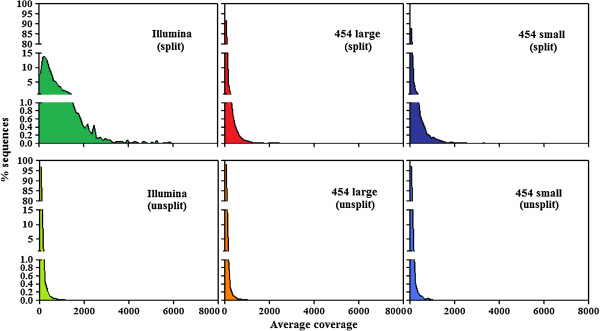
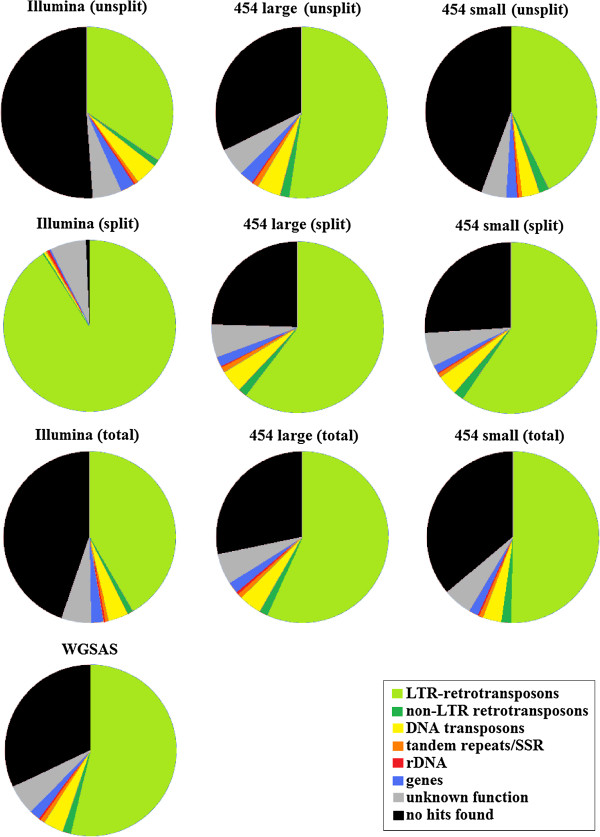
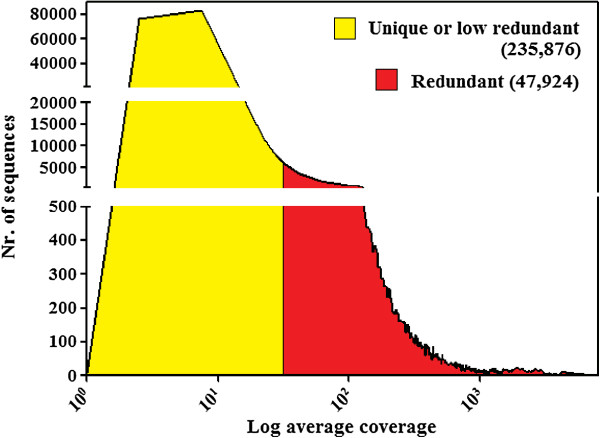
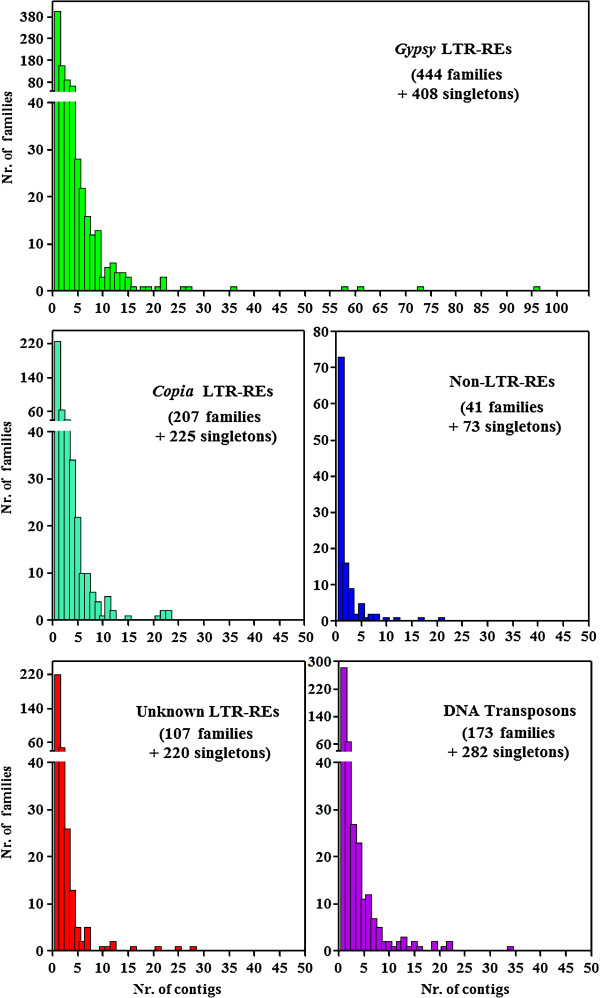
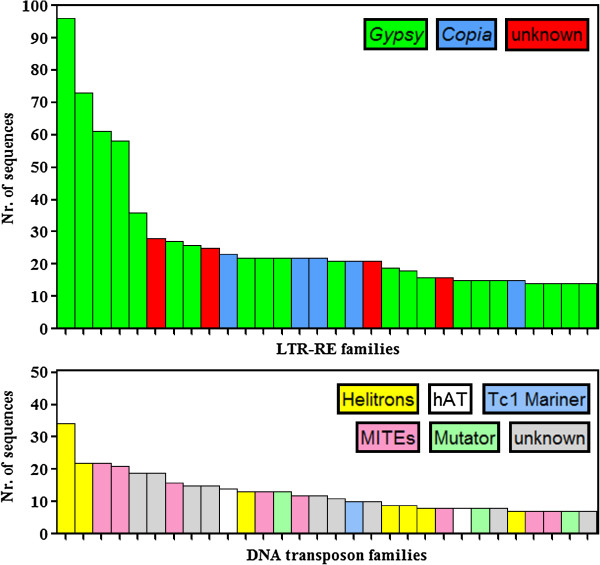
Results: By varying sequencing technology (Illumina or 454), coverage (0.55 x-1.25 x), assemblers and assembly procedures, six different genomic databases were produced. The annotation of these databases showed that they were composed of different proportions of repetitive DNA families. The final assembly of the sequences belonging to the six databases produced a whole genome set of 283,800 contigs. The redundancy of each contig was estimated by mapping the whole genome set with a large Illumina read set and measuring the number of matched Illumina reads. The repetitive component amounted to 81% of the sunflower genome, that is composed mainly of numerous families of Gypsy and Copia retrotransposons. Also many families of non autonomous retrotransposons and DNA transposons (especially of the Helitron superfamily) were identified.
Conclusions: The results substantially matched those previously obtained by using a Sanger-sequenced shotgun library and a standard 454 whole-genome-shotgun approach, indicating the reliability of the proposed procedures also for other species. The repetitive sequences were collected to produce a database, SUNREP, that will be useful for the annotation of the sunflower genome sequence and for studying the genome evolution in dicotyledons.
Figures
References
-
- Pellicer J, Fay MF, Leitch IJ. The largest eukaryotic genome of them all? Bot J Linnean Soc. 2010;164:10–15. doi: 10.1111/j.1095-8339.2010.01072.x. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
