

Genotype- and phenotype-guided management of congenital long QT syndrome
- PMID: 24093767
- PMCID: PMC3940076
- DOI: 10.1016/j.cpcardiol.2013.08.001
Genotype- and phenotype-guided management of congenital long QT syndrome
Abstract
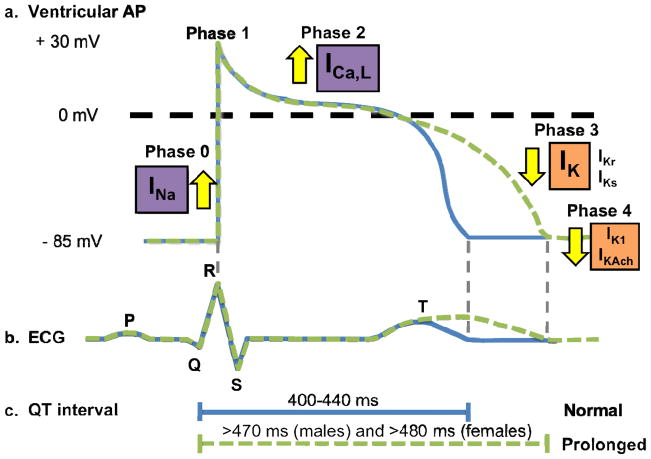
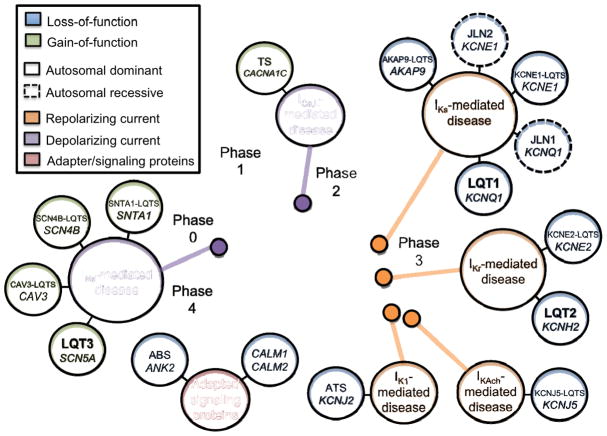
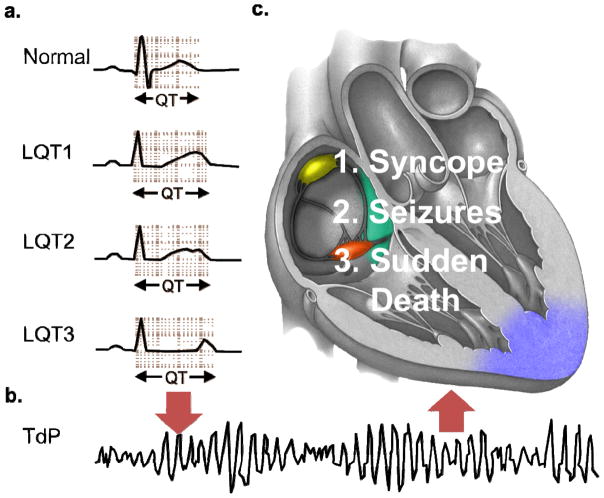
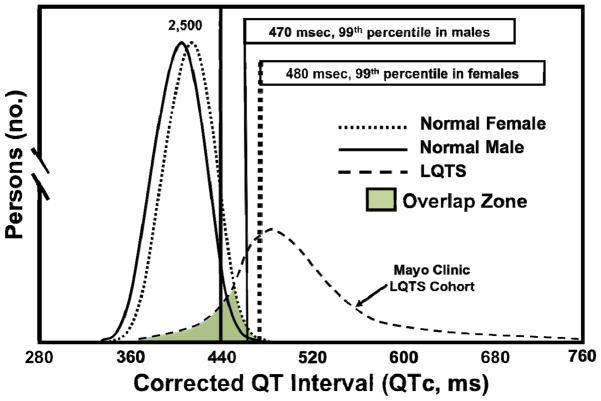
Congenital long QT syndrome (LQTS) is a genetically heterogeneous group of heritable disorders of myocardial repolarization linked by the shared clinical phenotype of QT prolongation on electrocardiogram and an increased risk of potentially life-threatening cardiac arrhythmias. At the molecular level, mutations in 15 distinct LQTS-susceptibility genes that encode ion channel pore-forming α-subunits and accessory β-subunits central to the electromechanical function of the heart have been implicated in its pathogenesis. Over the past 2 decades, our evolving understanding of the electrophysiological mechanisms by which specific genetic substrates perturb the cardiac action potential has translated into vastly improved approaches to the diagnosis, risk stratification, and treatment of patients with LQTS. In this review, we describe how our understanding of the molecular underpinnings of LQTS has yielded numerous clinically meaningful genotype-phenotype correlations and how these insights have translated into genotype- and phenotype-guided approaches to the clinical management of LQTS.
© 2013 Elsevier B.V. All rights reserved.
Figures
References
-
- Moss AJ. Long QT Syndrome. JAMA. 2003;289:2041–4. - PubMed
-
- Romano C, Gemme G, Pongiglione R. Rare Cardiac Arrythmias of the Pediatric Age. Ii. Syncopal Attacks Due to Paroxysmal Ventricular Fibrillation. (Presentation of 1st Case in Italian Pediatric Literature) Clin Pediatr (Bologna) 1963;45:656–83. - PubMed
-
- Ward OC. A New Familial Cardiac Syndrome in Children. J Ir Med Assoc. 1964;54:103–6. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
