

Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK
- PMID: 24095877
- PMCID: PMC3896127
- DOI: 10.1016/j.yjmcc.2013.09.015
Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK
Abstract
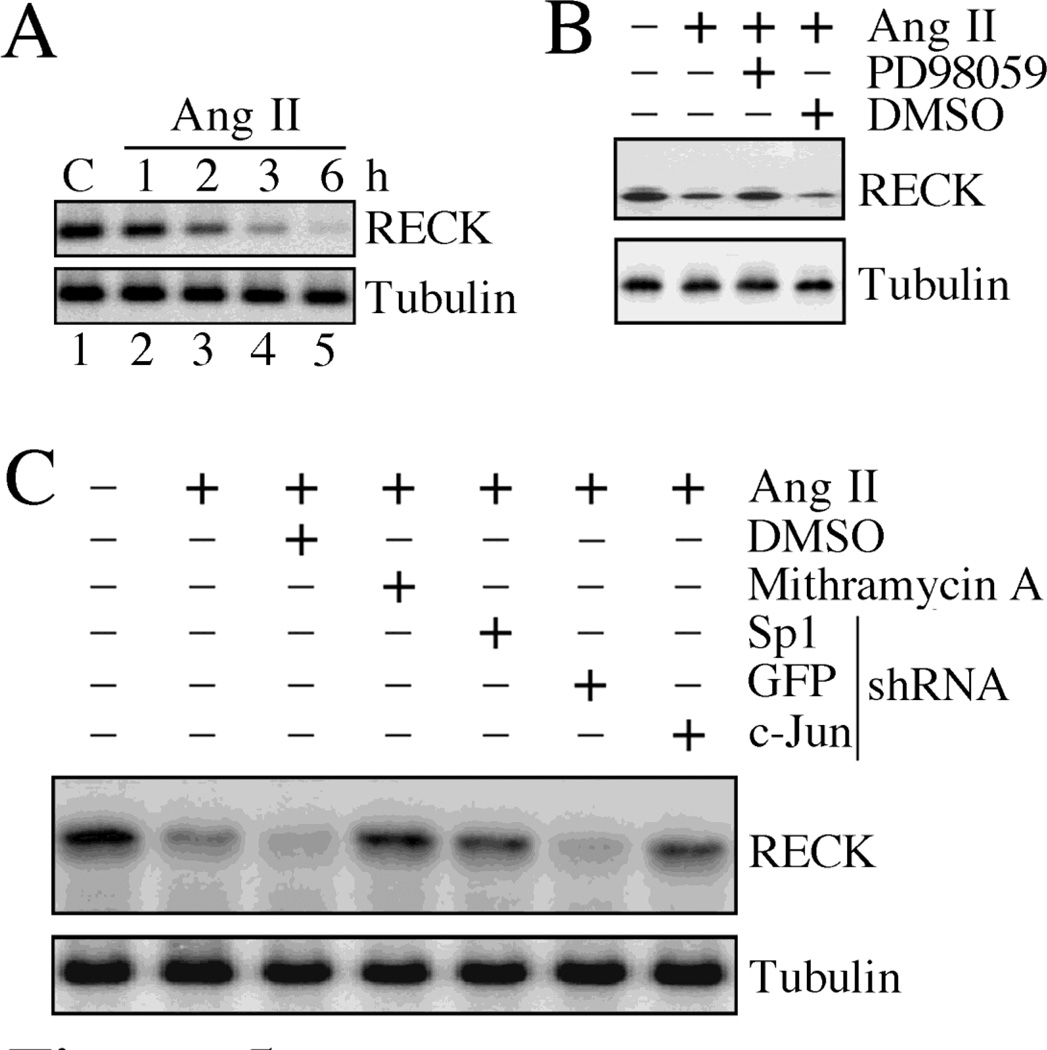
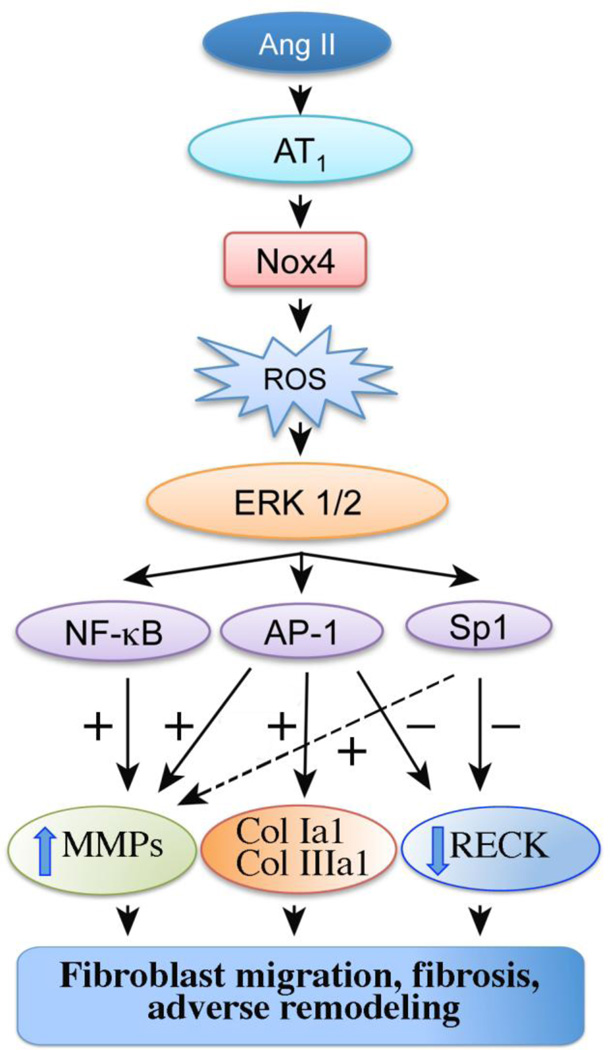
Sustained induction and activation of matrixins (matrix metalloproteinases or MMPs), and the destruction and deposition of extracellular matrix (ECM), are the hallmarks of cardiac fibrosis. The reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) is a unique membrane-anchored endogenous MMP regulator. We hypothesized that elevated angiotensin II (Ang II), which is associated with fibrosis in the heart, differentially regulates MMPs and RECK both in vivo and in vitro. Continuous infusion of Ang II into male C57Bl/6 mice for 2weeks resulted in cardiac fibrosis, with increased expressions of MMPs 2, 7, 9 and 14, and of collagens Ia1 and IIIa1. The expression of RECK, however, was markedly suppressed. These effects were inhibited by co-treatment with the Ang II type 1 receptor (AT1) antagonist losartan. In vitro, Ang II suppressed RECK expression in adult mouse cardiac fibroblasts (CF) via AT1/Nox4-dependent ERK/Sp1 activation, but induced MMPs 2, 14 and 9 via NF-κB, AP-1 and/or Sp1 activation. Further, while forced expression of RECK inhibits, its knockdown potentiates Ang II-induced CF migration. Notably, RECK overexpression reduced Ang II-induced MMPs 2, 9 and 14 activation, but enhanced collagens Ia1 and IIIa1 expression and soluble collagen release. These results demonstrate for the first time that Ang II suppresses RECK, but induces MMPs both in vivo and in vitro, and RECK overexpression blunts Ang II-induced MMP activation and CF migration in vitro. Strategies that upregulate RECK expression in vivo have the potential to attenuate sustained MMP expression, and blunt fibrosis and adverse remodeling in hypertensive heart diseases.
Keywords: 12-O-tetradecanoylphorbol-13-acetate; 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide; ADAM; AP-1; ARB; AT1; AT2; Adverse remodeling; CF; CMV; CVD; Cardiac fibrosis; DPI; ECM; EGFP; ERK; Elk; Ets-like protein; GAPDH; GFP; GPI; JNK; LV; MAPK; MMP; MOI; MT1-MMP; MTT; NADPH; NADPH oxidase; NF-κB; Nox; Nox4; RECK; ROS; SBP; Sp1; TACE; TIMP; TPA; TPA DNA response element; TRE; UTR; WT; a disintegrin and metalloproteinase domain; activator protein-1; angiotensin II type 1 receptor; angiotensin II type II receptor; angiotensin receptor blockers; c-Jun amino-terminal kinase; cardiac fibroblasts; cardiovascular disease; cytomegalovirus; diphenylene iodonium; enhanced green fluorescent protein; extracellular matrix; extracellular signal-regulated kinase; glyceraldehyde-3-phosphate dehydrogenase; glycophosphatidylinositol; green fluorescent protein; left ventricle; matrix metalloproteinase; membrane type 1-MMP; mitogen-activated protein kinase; multiplicity of infection; nicotinamide adenine dinucleotide phosphate; nuclear factor kappa B; reactive oxygen species; reversion-inducing-cysteine-rich protein with Kazal motifs; shRNA; short hairpin RNA; siMMP; small inhibitory RNA against MMP; specific protein 1; systolic blood pressure; tissue inhibitor of metalloproteinase; tumor necrosis factor, alpha, converting enzyme; untranslated region; wild-type.
© 2013.
Conflict of interest statement
Figures






References
-
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000 Sep;52(3):415–472. - PubMed
-
- Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009 Aug;123(2):255–278. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
