

GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis
- PMID: 24096415
- PMCID: PMC3837814
- DOI: 10.1128/AEM.02411-13
GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis
Abstract
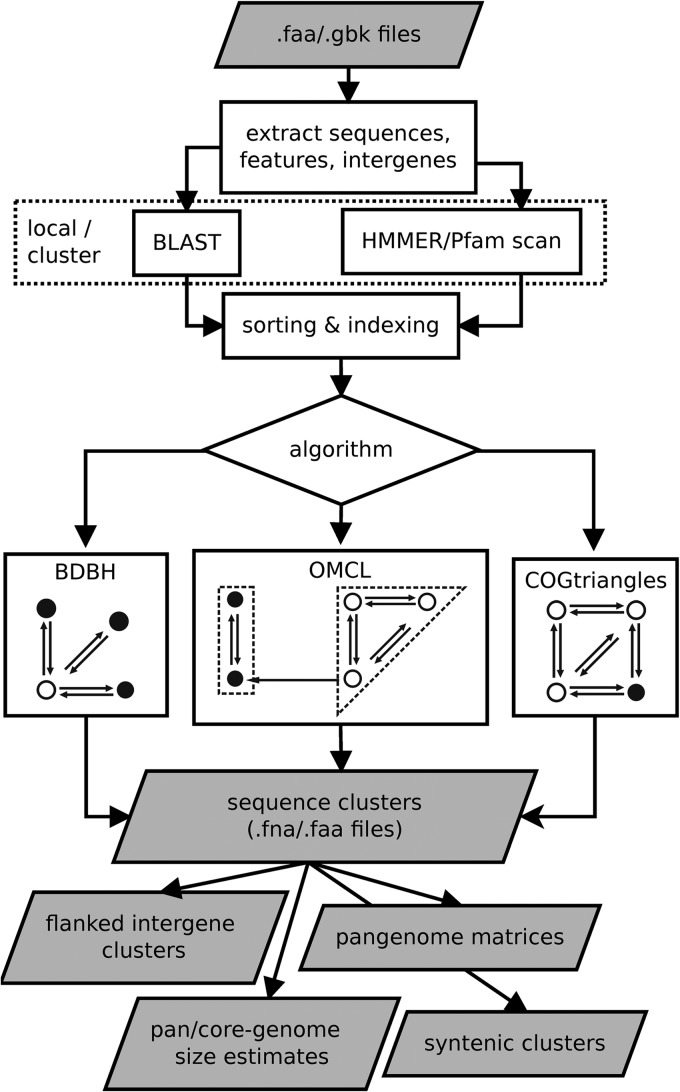
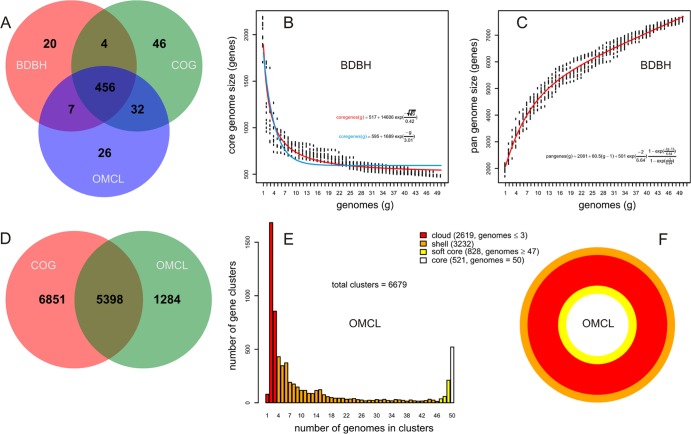
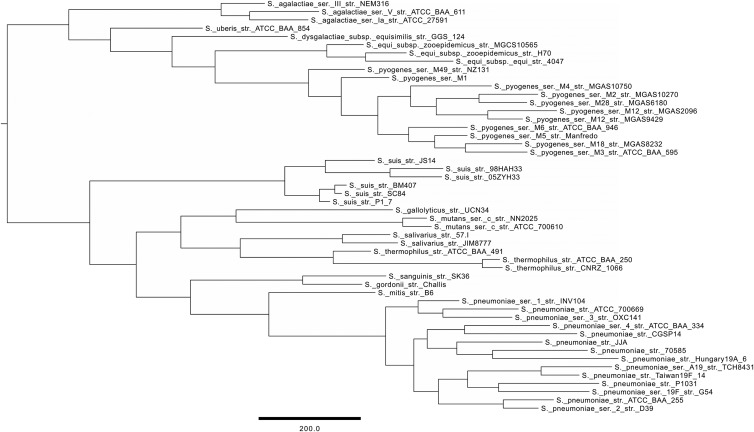
GET_HOMOLOGUES is an open-source software package that builds on popular orthology-calling approaches making highly customizable and detailed pangenome analyses of microorganisms accessible to nonbioinformaticians. It can cluster homologous gene families using the bidirectional best-hit, COGtriangles, or OrthoMCL clustering algorithms. Clustering stringency can be adjusted by scanning the domain composition of proteins using the HMMER3 package, by imposing desired pairwise alignment coverage cutoffs, or by selecting only syntenic genes. The resulting homologous gene families can be made even more robust by computing consensus clusters from those generated by any combination of the clustering algorithms and filtering criteria. Auxiliary scripts make the construction, interrogation, and graphical display of core genome and pangenome sets easy to perform. Exponential and binomial mixture models can be fitted to the data to estimate theoretical core genome and pangenome sizes, and high-quality graphics can be generated. Furthermore, pangenome trees can be easily computed and basic comparative genomics performed to identify lineage-specific genes or gene family expansions. The software is designed to take advantage of modern multiprocessor personal computers as well as computer clusters to parallelize time-consuming tasks. To demonstrate some of these capabilities, we survey a set of 50 Streptococcus genomes annotated in the Orthologous Matrix (OMA) browser as a benchmark case. The package can be downloaded at http://www.eead.csic.es/compbio/soft/gethoms.php and http://maya.ccg.unam.mx/soft/gethoms.php.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
