

Insulin resistance protects the heart from fuel overload in dysregulated metabolic states
- PMID: 24097426
- PMCID: PMC3882545
- DOI: 10.1152/ajpheart.00854.2012
Insulin resistance protects the heart from fuel overload in dysregulated metabolic states
Abstract
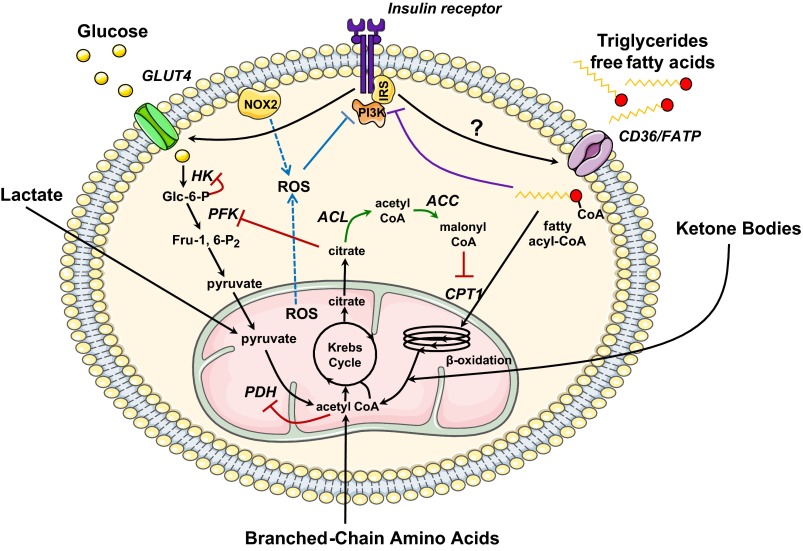
Reversing impaired insulin sensitivity has been suggested as treatment for heart failure. However, recent clinical evidence suggests the opposite. Here we present a line of reasoning in support of the hypothesis that insulin resistance protects the heart from the consequences of fuel overload in the dysregulated metabolic state of obesity and diabetes. We discuss pathways of myocardial fuel toxicity, as well as several layers of defense against fuel overload. Our reassessment of the literature suggests that in the heart, insulin-sensitizing agents result in an elimination of some of the defenses, leading to cytotoxic damage. In contrast, a normalization of fuel supply should either prevent or reverse the process. Taken together, we offer a new perspective on insulin resistance of the heart.
Keywords: insulin resistance; metabolism; obesity.
Figures
References
-
- Anker SD, von Haehling S. The obesity paradox in heart failure: accepting reality and making rational decisions. Clin Pharmacol Ther 90: 188–190, 2011 - PubMed
-
- Augustus AS, Buchanan J, Park TS, Hirata K, Noh HL, Sun J, Homma S, D'Armiento J, Abel ED, Goldberg IJ. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J Biol Chem 281: 8716–8723, 2006 - PubMed
-
- Balteau M, Tajeddine N, de Meester C, Ginion A, Des Rosiers C, Brady NR, Sommereyns C, Horman S, Vanoverschelde JL, Gailly P, Hue L, Bertrand L, Beauloye C. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res 92: 237–246, 2011 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical