

Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development
- PMID: 24098934
- PMCID: PMC4053753
- DOI: 10.1186/gb-2013-14-10-r108
Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development
Abstract
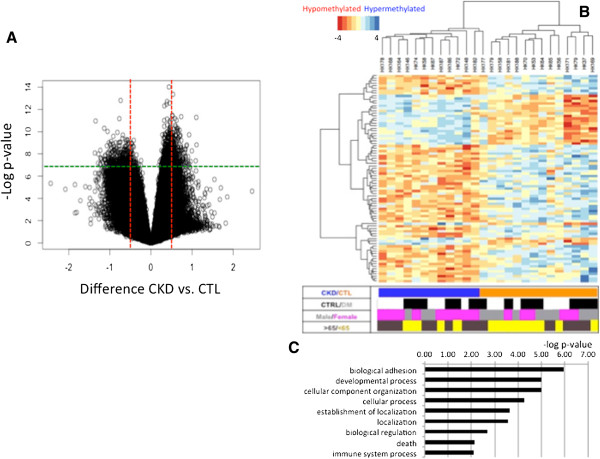
Background: One in eleven people is affected by chronic kidney disease, a condition characterized by kidney fibrosis and progressive loss of kidney function. Epidemiological studies indicate that adverse intrauterine and postnatal environments have a long-lasting role in chronic kidney disease development. Epigenetic information represents a plausible carrier for mediating this programming effect. Here we demonstrate that genome-wide cytosine methylation patterns of healthy and chronic kidney disease tubule samples obtained from patients show significant differences.
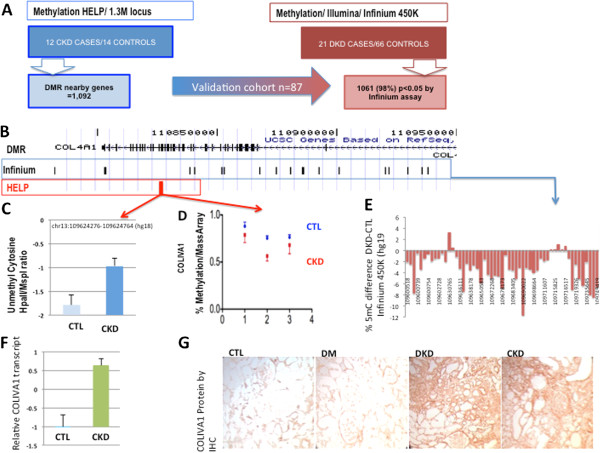
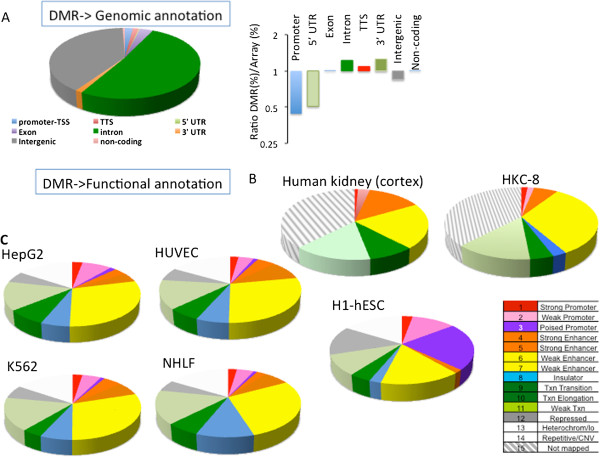
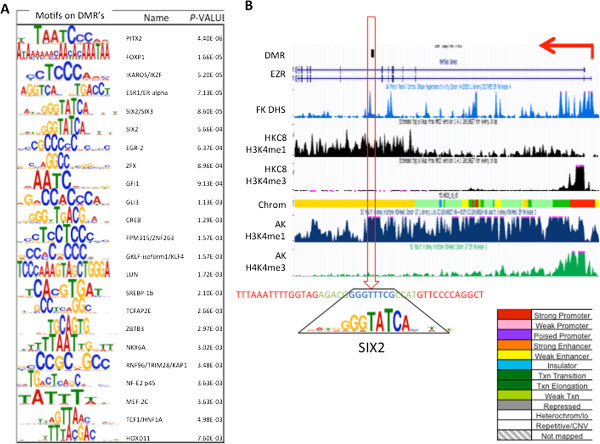
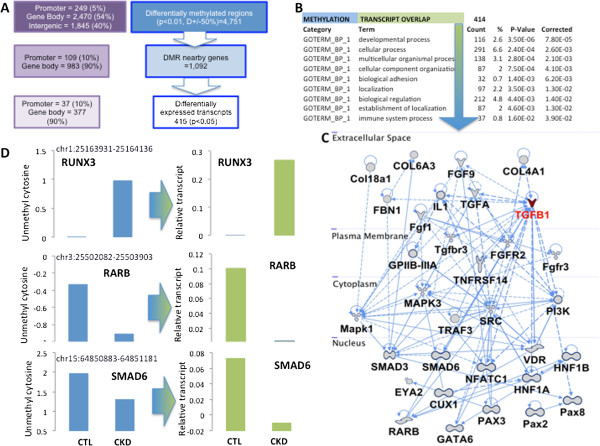
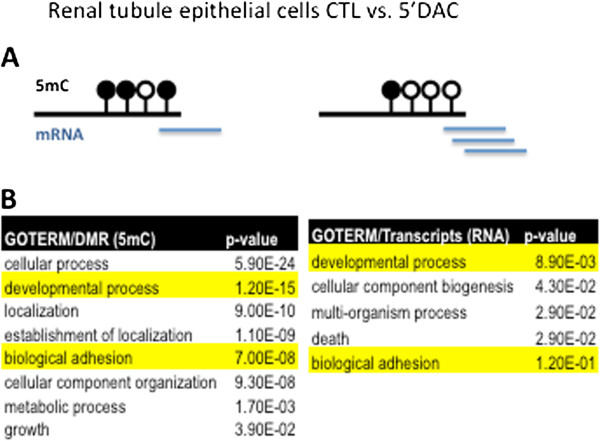
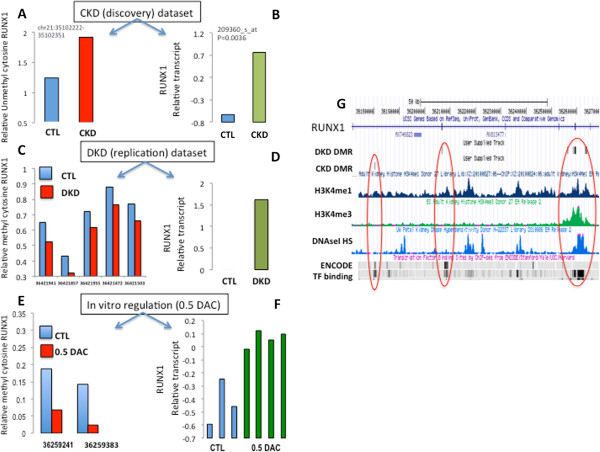
Results: We identify differentially methylated regions and validate these in a large replication dataset. The differentially methylated regions are rarely observed on promoters, but mostly overlap with putative enhancer regions, and they are enriched in consensus binding sequences for important renal transcription factors. This indicates their importance in gene expression regulation. A core set of genes that are known to be related to kidney fibrosis, including genes encoding collagens, show cytosine methylation changes correlating with downstream transcript levels.
Conclusions: Our report raises the possibility that epigenetic dysregulation plays a role in chronic kidney disease development via influencing core pro-fibrotic pathways and can aid the development of novel biomarkers and future therapeutics.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
