

Oxidative stress in the pathology and treatment of systemic lupus erythematosus
- PMID: 24100461
- PMCID: PMC4046645
- DOI: 10.1038/nrrheum.2013.147
Oxidative stress in the pathology and treatment of systemic lupus erythematosus
Abstract
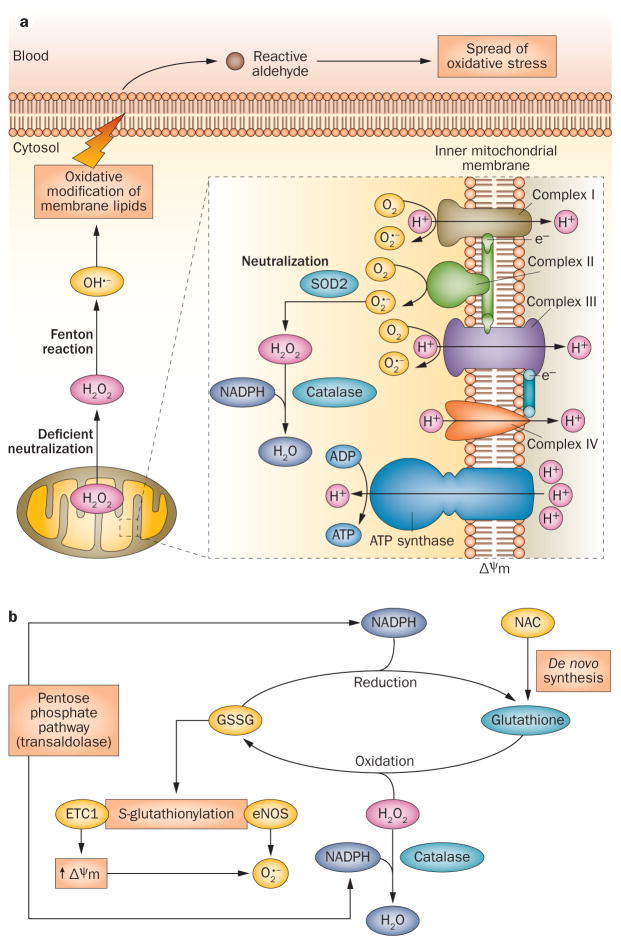
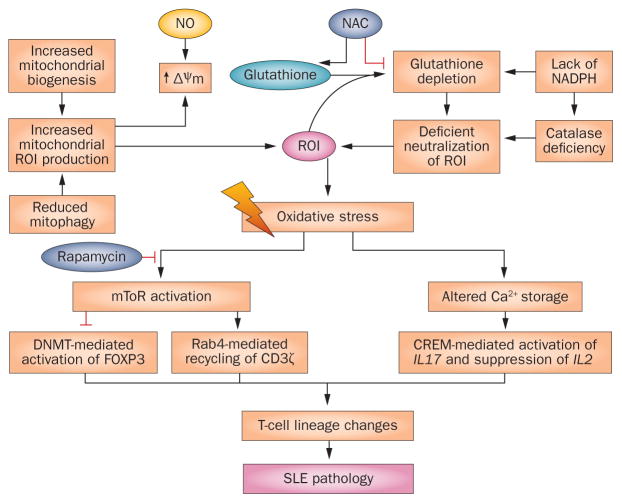
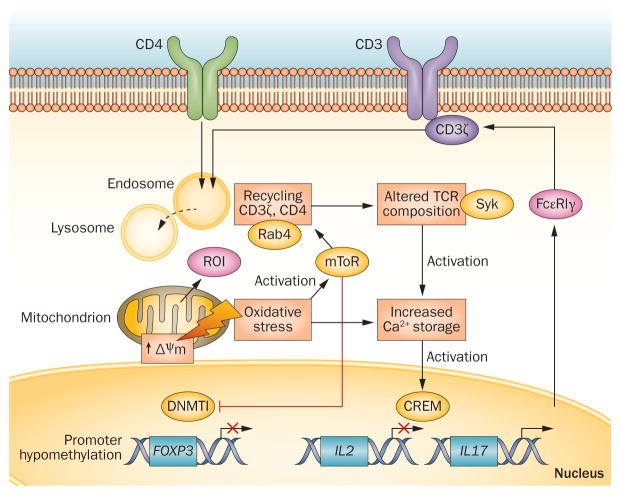
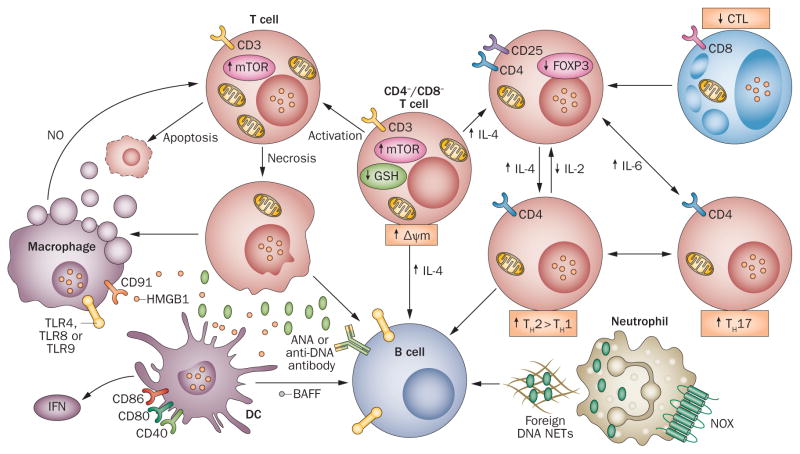
Oxidative stress is increased in systemic lupus erythematosus (SLE), and it contributes to immune system dysregulation, abnormal activation and processing of cell-death signals, autoantibody production and fatal comorbidities. Mitochondrial dysfunction in T cells promotes the release of highly diffusible inflammatory lipid hydroperoxides, which spread oxidative stress to other intracellular organelles and through the bloodstream. Oxidative modification of self antigens triggers autoimmunity, and the degree of such modification of serum proteins shows striking correlation with disease activity and organ damage in SLE. In T cells from patients with SLE and animal models of the disease, glutathione, the main intracellular antioxidant, is depleted and serine/threonine-protein kinase mTOR undergoes redox-dependent activation. In turn, reversal of glutathione depletion by application of its amino acid precursor, N-acetylcysteine, improves disease activity in lupus-prone mice; pilot studies in patients with SLE have yielded positive results that warrant further research. Blocking mTOR activation in T cells could conceivably provide a well-tolerated and inexpensive alternative approach to B-cell blockade and traditional immunosuppressive treatments. Nevertheless, compartmentalized oxidative stress in self-reactive T cells, B cells and phagocytic cells might serve to limit autoimmunity and its inhibition could be detrimental. Antioxidant therapy might also be useful in ameliorating damage caused by other treatments. This Review thus seeks to critically evaluate the complexity of oxidative stress and its relevance to the pathogenesis and treatment of SLE.
Conflict of interest statement
The author declares no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
