

Indistinguishable synaptic pharmacodynamics of the N-methyl-D-aspartate receptor channel blockers memantine and ketamine
- PMID: 24101301
- PMCID: PMC3834148
- DOI: 10.1124/mol.113.089334
Indistinguishable synaptic pharmacodynamics of the N-methyl-D-aspartate receptor channel blockers memantine and ketamine
Abstract
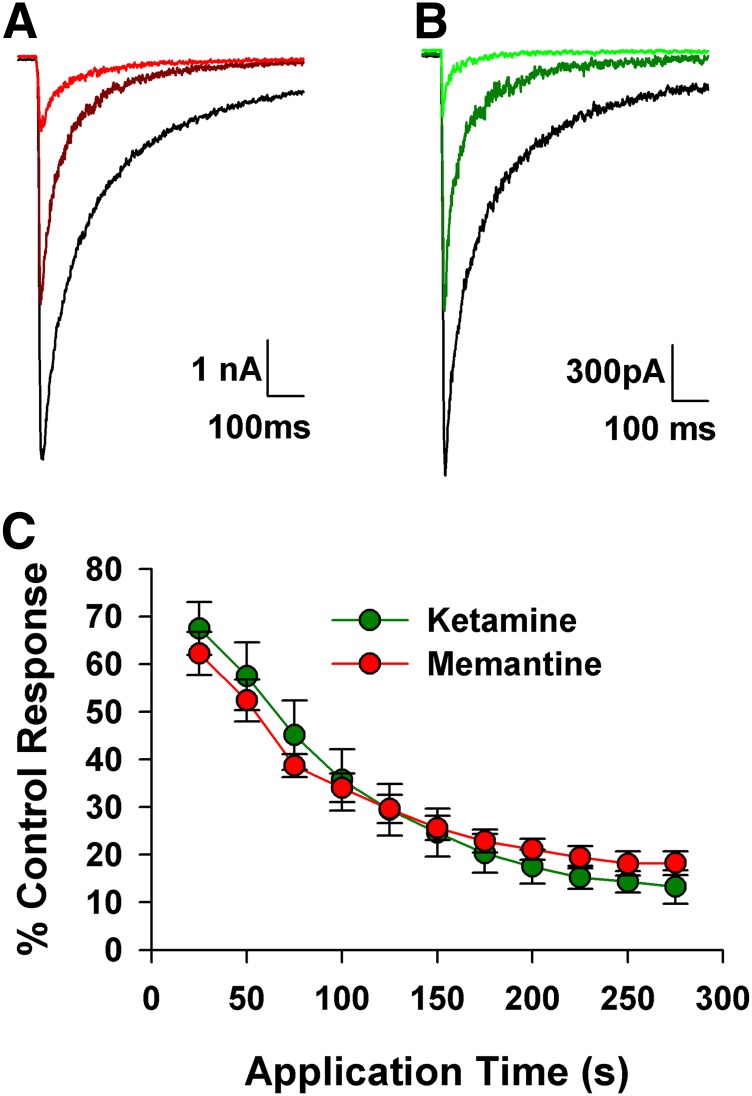
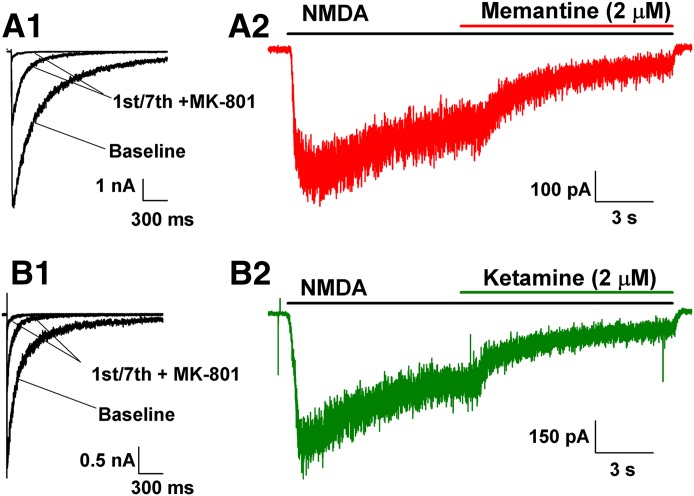
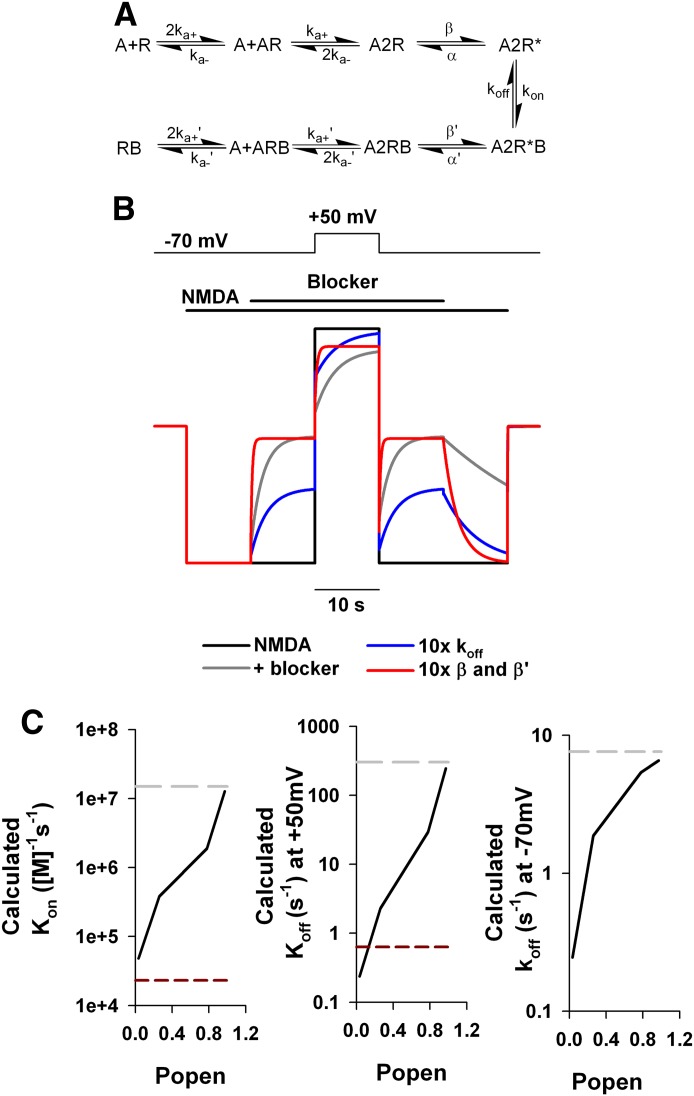
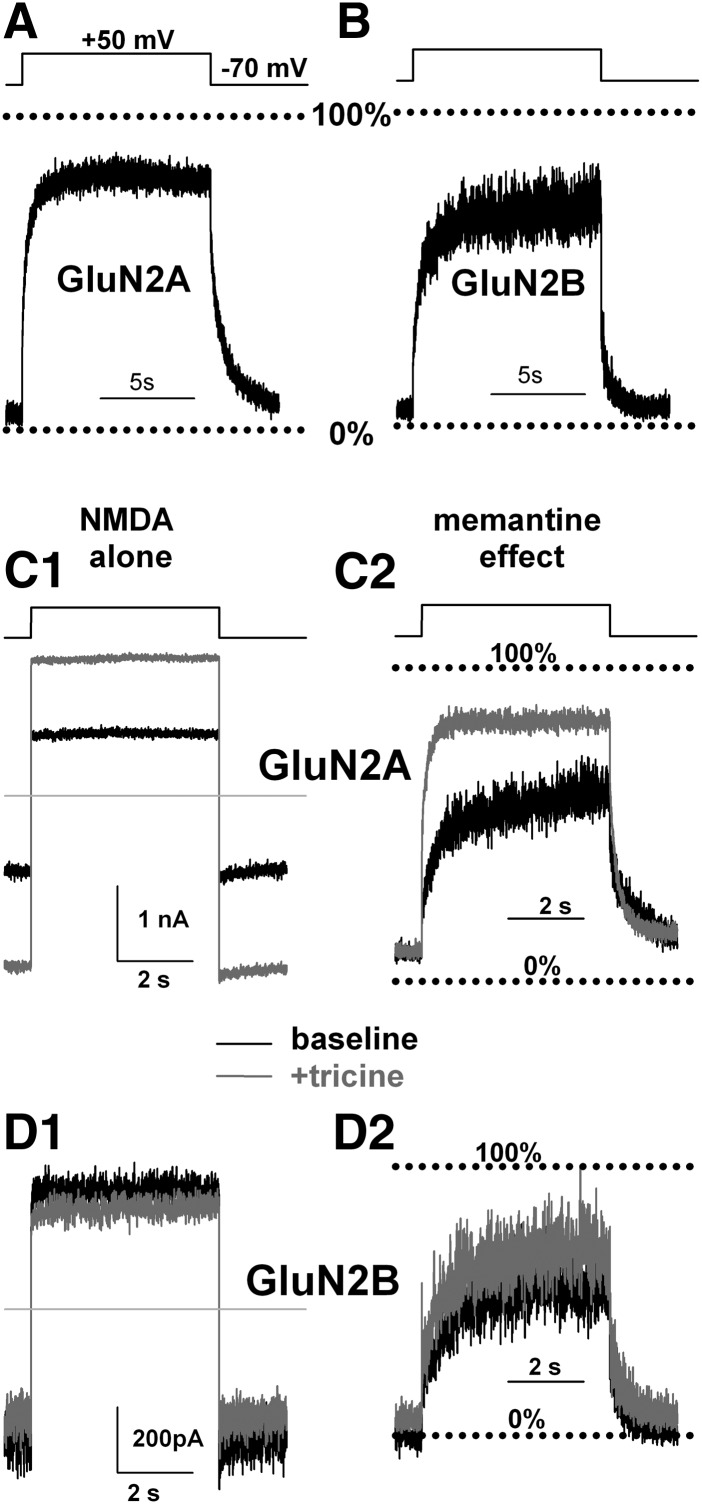
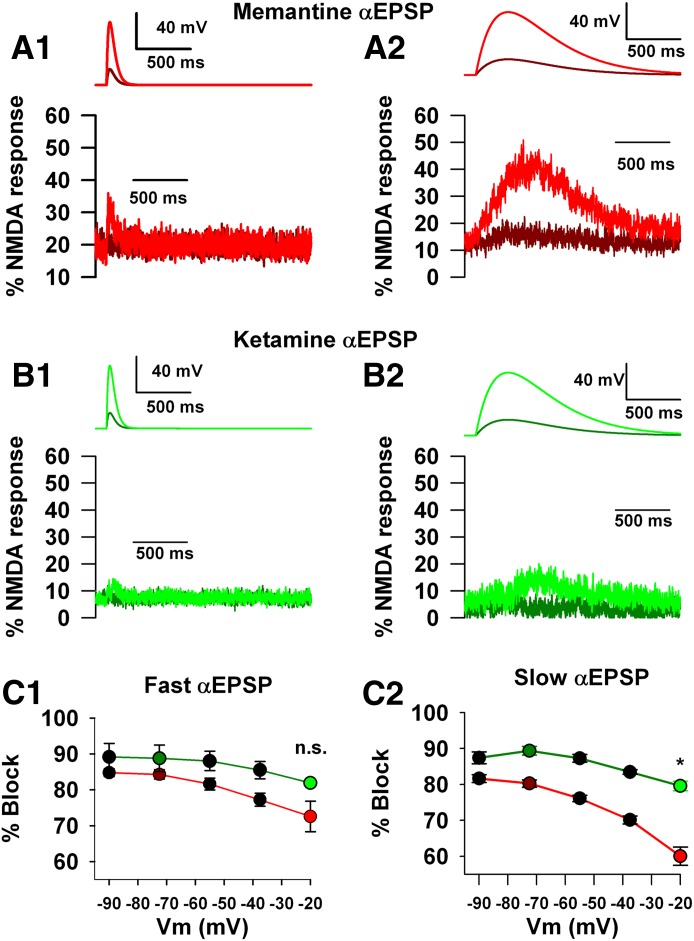
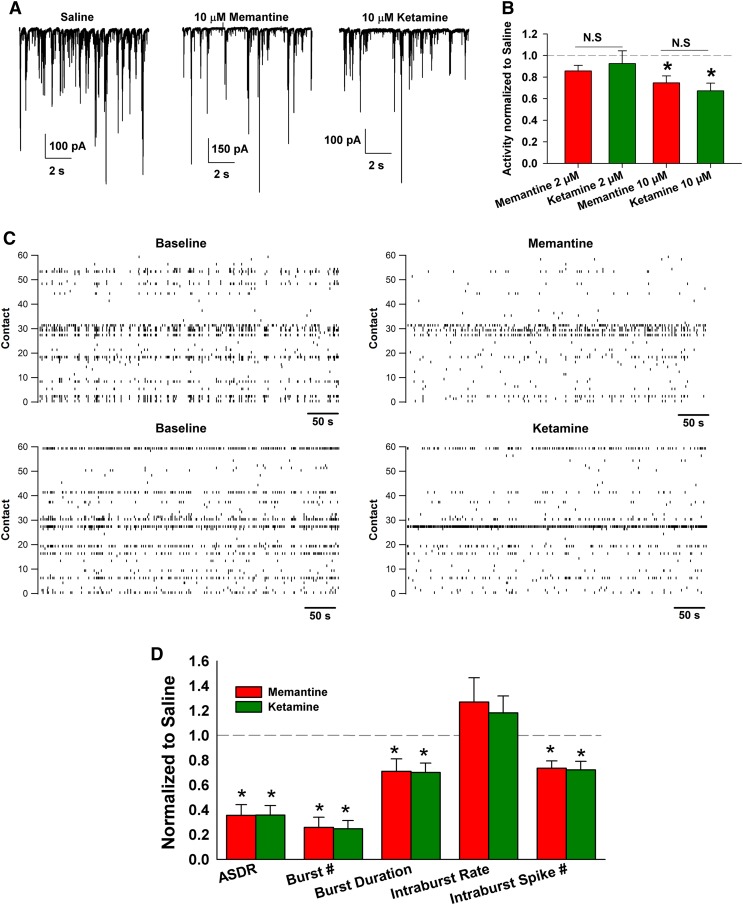
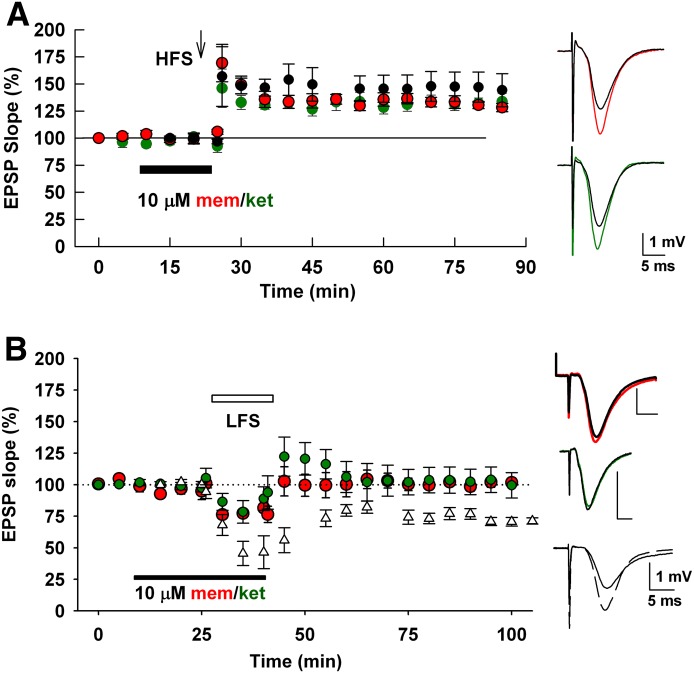
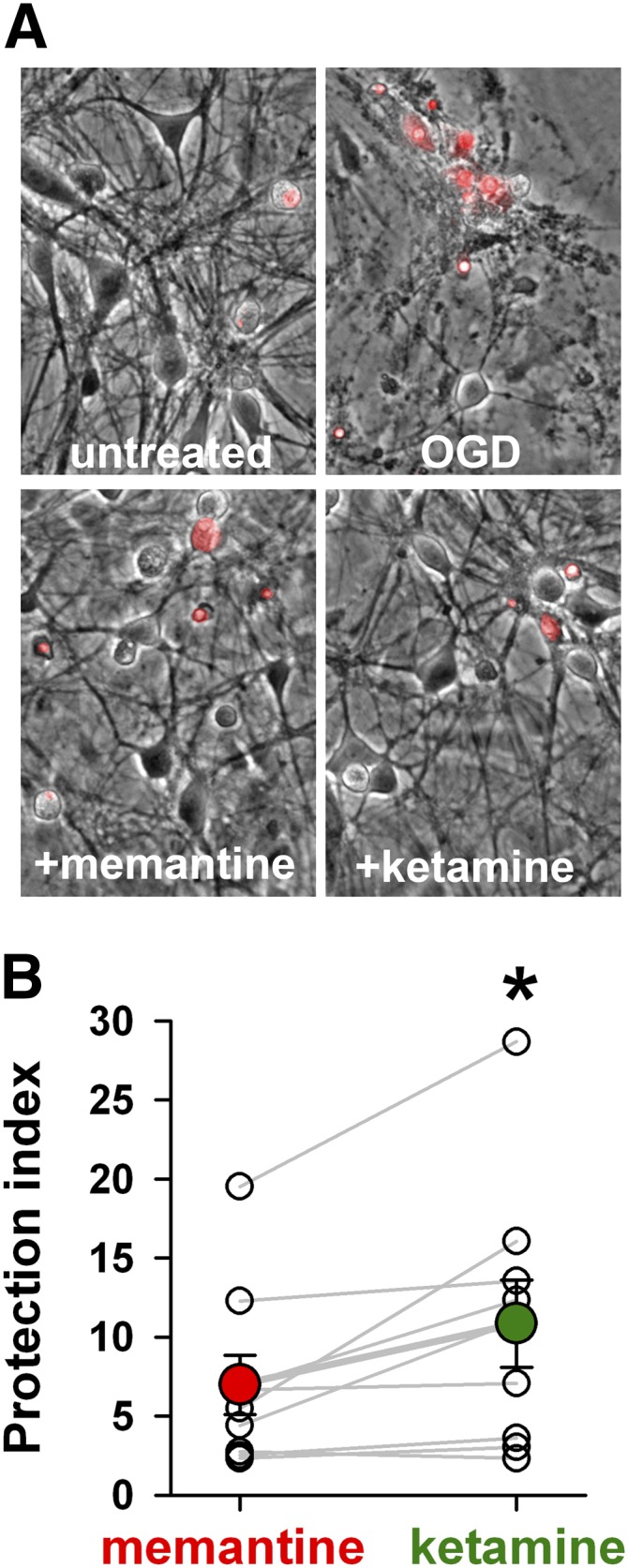
Memantine and ketamine, voltage- and activation-dependent channel blockers of N-methyl-d-aspartate (NMDA) receptors (NMDARs), have enjoyed a recent resurgence in clinical interest. Steady-state pharmacodynamic differences between these blockers have been reported, but it is unclear whether the compounds differentially affect dynamic physiologic signaling. In this study, we explored nonequilibrium conditions relevant to synaptic transmission in hippocampal networks in dissociated culture and hippocampal slices. Equimolar memantine and ketamine had indistinguishable effects on the following measures: steady-state NMDA currents, NMDAR excitatory postsynaptic current (EPSC) decay kinetics, progressive EPSC inhibition during repetitive stimulation, and extrasynaptic NMDAR inhibition. Therapeutic drug efficacy and tolerability of memantine have been attributed to fast kinetics and strong voltage dependence. However, pulse depolarization in drug presence revealed a surprisingly slow and similar time course of equilibration for the two compounds, although memantine produced a more prominent fast component (62% versus 48%) of re-equilibration. Simulations predicted that low gating efficacy underlies the slow voltage-dependent relief from block. This prediction was empirically supported by faster voltage-dependent blocker re-equilibration with several experimental manipulations of gating efficacy. Excitatory postsynaptic potential-like voltage commands produced drug differences only with large, prolonged depolarizations unlikely to be attained physiologically. In fact, we found no difference between drugs on measures of spontaneous network activity or acute effects on plasticity in hippocampal slices. Despite indistinguishable synaptic pharmacodynamics, ketamine provided significantly greater neuroprotection from damage induced by oxygen glucose deprivation, consistent with the idea that under extreme depolarizing conditions, the biophysical difference between drugs becomes detectable. We conclude that despite subtle differences in voltage dependence, during physiologic activity, blocker pharmacodynamics are largely indistinguishable and largely voltage independent.
Figures



Similar articles
-
Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses.J Neurosci. 2010 Aug 18;30(33):11246-50. doi: 10.1523/JNEUROSCI.2488-10.2010. J Neurosci. 2010. PMID: 20720132 Free PMC article.
-
Memantine and Ketamine Differentially Alter NMDA Receptor Desensitization.J Neurosci. 2017 Oct 4;37(40):9686-9704. doi: 10.1523/JNEUROSCI.1173-17.2017. Epub 2017 Sep 6. J Neurosci. 2017. PMID: 28877967 Free PMC article.
-
Interaction between positive allosteric modulators and trapping blockers of the NMDA receptor channel.Br J Pharmacol. 2015 Mar;172(5):1333-47. doi: 10.1111/bph.13007. Epub 2015 Jan 13. Br J Pharmacol. 2015. PMID: 25377730 Free PMC article.
-
The neuropharmacological basis for the use of memantine in the treatment of Alzheimer's disease.CNS Drug Rev. 2003 Fall;9(3):275-308. doi: 10.1111/j.1527-3458.2003.tb00254.x. CNS Drug Rev. 2003. PMID: 14530799 Free PMC article. Review.
-
Mechanism of action of memantine.Curr Opin Pharmacol. 2006 Feb;6(1):61-7. doi: 10.1016/j.coph.2005.09.007. Epub 2005 Dec 20. Curr Opin Pharmacol. 2006. PMID: 16368266 Review.
Cited by
-
Ketamine and phencyclidine: the good, the bad and the unexpected.Br J Pharmacol. 2015 Sep;172(17):4254-76. doi: 10.1111/bph.13222. Epub 2015 Jul 28. Br J Pharmacol. 2015. PMID: 26075331 Free PMC article. Review.
-
Uncovering the Underlying Mechanisms of Ketamine as a Novel Antidepressant.Front Pharmacol. 2022 Jul 7;12:740996. doi: 10.3389/fphar.2021.740996. eCollection 2021. Front Pharmacol. 2022. PMID: 35872836 Free PMC article. Review.
-
Imprecision in Precision Medicine: Differential Response of a Disease-Linked GluN2A Mutant to NMDA Channel Blockers.Front Pharmacol. 2021 Oct 28;12:773455. doi: 10.3389/fphar.2021.773455. eCollection 2021. Front Pharmacol. 2021. PMID: 34776984 Free PMC article.
-
Ketamine: promising path or false prophecy in the development of novel therapeutics for mood disorders?Neuropsychopharmacology. 2015 Jan;40(2):259-67. doi: 10.1038/npp.2014.261. Epub 2014 Sep 26. Neuropsychopharmacology. 2015. PMID: 25257213 Free PMC article. Review.
-
Duality of Antidepressants and Neuroprotectants.Neurotox Res. 2016 Jul;30(1):1-13. doi: 10.1007/s12640-015-9577-1. Epub 2015 Nov 27. Neurotox Res. 2016. PMID: 26613895 Free PMC article. Review.
References
-
- Bear MF. (1995) Mechanism for a sliding synaptic modification threshold. Neuron 15:1–4 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
