

Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction
- PMID: 24103571
- PMCID: PMC3835263
- DOI: 10.1161/JAHA.113.000461
Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction
Abstract
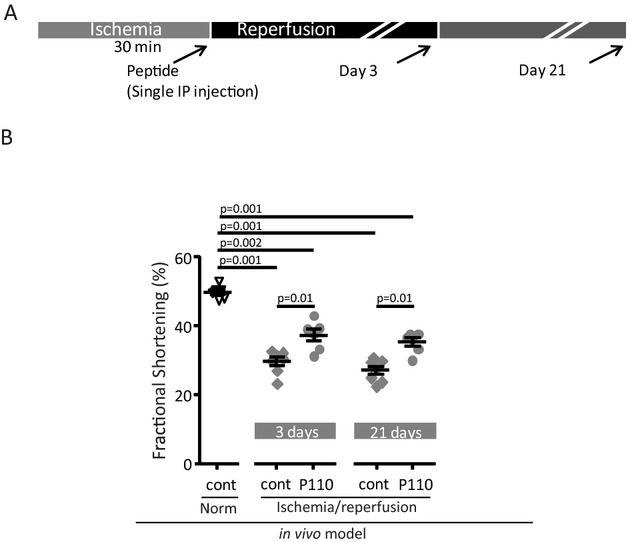
Background: Ischemia and reperfusion (IR) injury remains a major cause of morbidity and mortality and multiple molecular and cellular pathways have been implicated in this injury. We determined whether acute inhibition of excessive mitochondrial fission at the onset of reperfusion improves mitochondrial dysfunction and cardiac contractility postmyocardial infarction in rats.
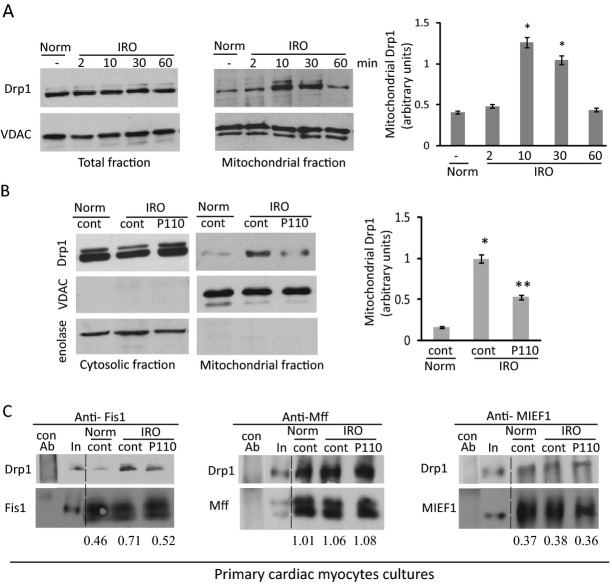
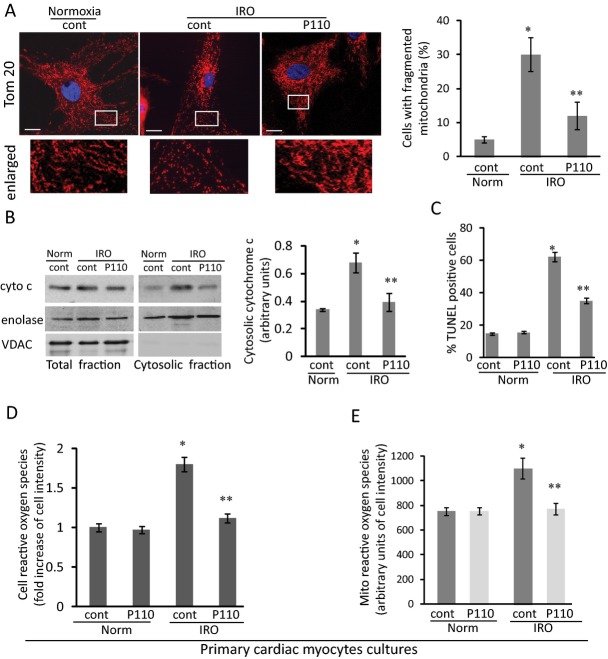
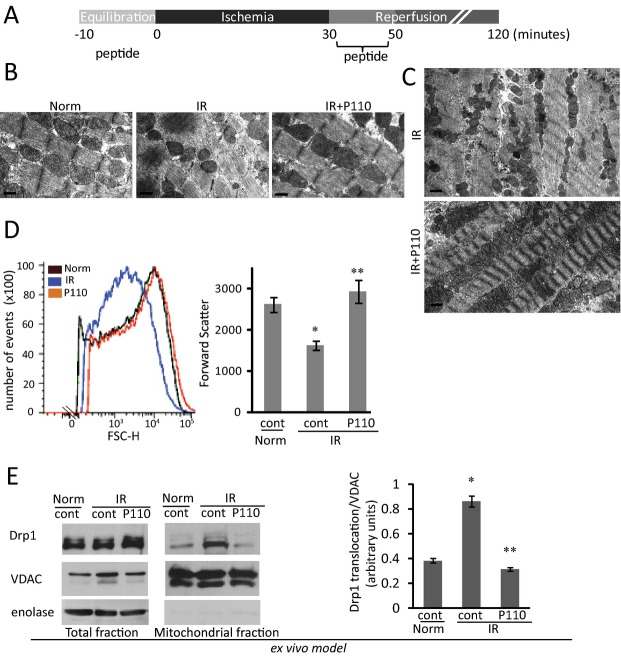
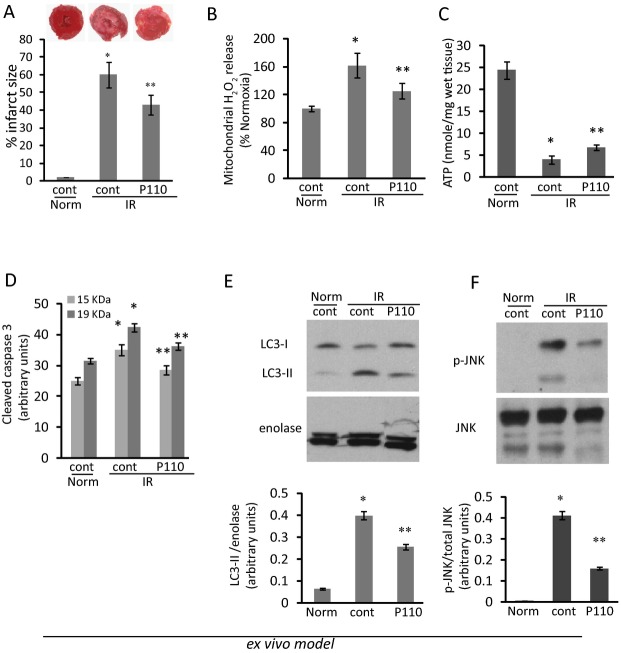
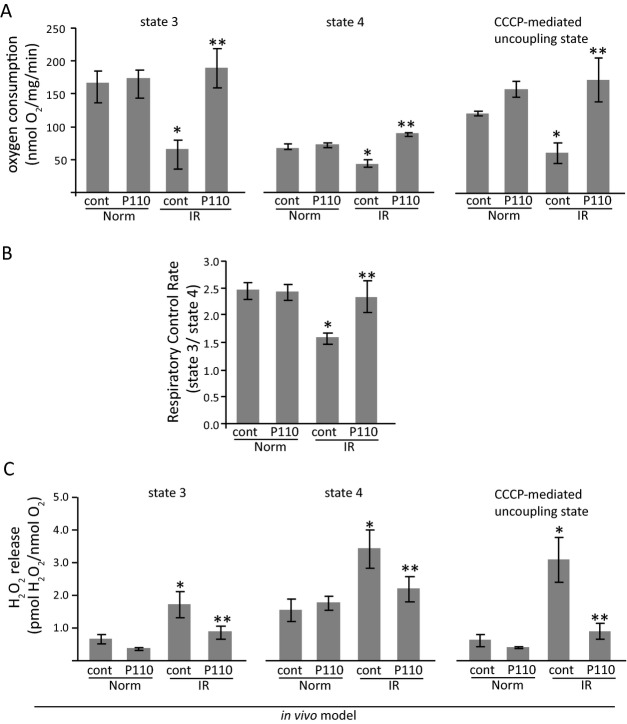
Methods and results: We used a selective inhibitor of the fission machinery, P110, which we have recently designed. P110 treatment inhibited the interaction of fission proteins Fis1/Drp1, decreased mitochondrial fission, and improved bioenergetics in three different rat models of IR, including primary cardiomyocytes, ex vivo heart model, and an in vivo myocardial infarction model. Drp1 transiently bound to the mitochondria following IR injury and P110 treatment blocked this Drp1 mitochondrial association. Compared with control treatment, P110 (1 μmol/L) decreased infarct size by 28 ± 2% and increased adenosine triphosphate levels by 70+1% after IR relative to control IR in the ex vivo model. Intraperitoneal injection of P110 (0.5 mg/kg) at the onset of reperfusion in an in vivo model resulted in improved mitochondrial oxygen consumption by 68% when measured 3 weeks after ischemic injury, improved cardiac fractional shortening by 35%, reduced mitochondrial H2O2 uncoupling state by 70%, and improved overall mitochondrial functions.
Conclusions: Together, we show that excessive mitochondrial fission at reperfusion contributes to long-term cardiac dysfunction in rats and that acute inhibition of excessive mitochondrial fission at the onset of reperfusion is sufficient to result in long-term benefits as evidenced by inhibiting cardiac dysfunction 3 weeks after acute myocardial infarction.
Keywords: Drp1; cardiac myocytes; heart; mitochondria; protein‐protein interaction inhibitor.
Figures
References
-
- Tandler B, Hoppel CL. Possible division of cardiac mitochondria. Anat Rec. 1972; 173:309-323 - PubMed
-
- Stanley WC, Hoppel CL. Mitochondrial dysfunction in heart failure: potential for therapeutic interventions? Cardiovasc Res. 2000; 45:805-806 - PubMed
-
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001; 33:1065-1089 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
