

The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells
- PMID: 24105369
- PMCID: PMC3992571
- DOI: 10.1038/ejhg.2013.209
The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells
Abstract
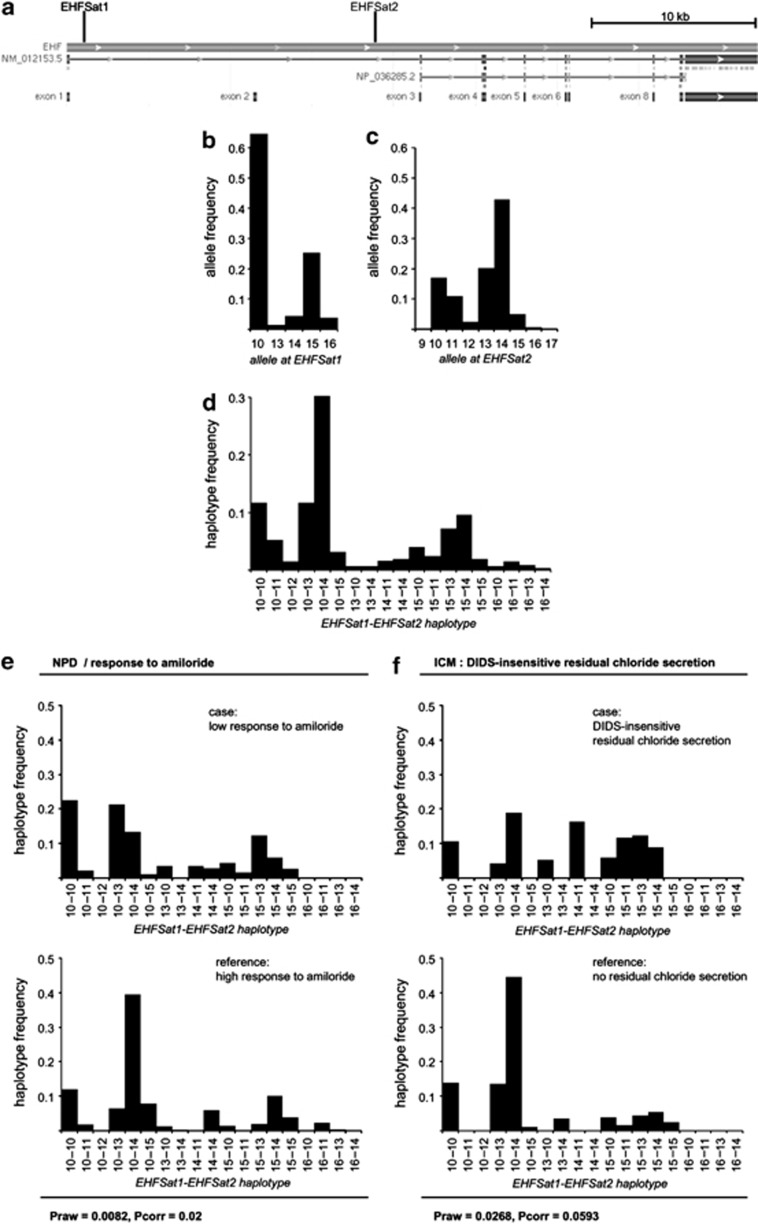
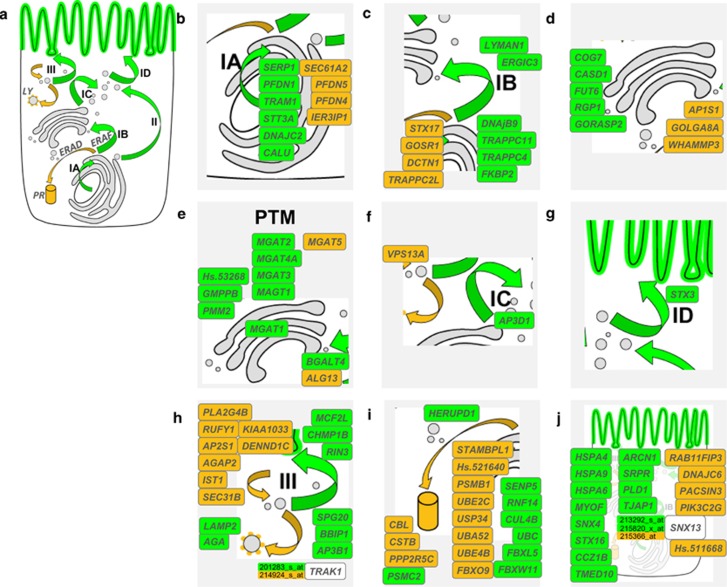
The three-base-pair deletion c.1521_1523delCTT (p.Phe508del, F508del) in the cystic fibrosis transmembrane conductance regulator (CFTR) is the most frequent disease-causing lesion in cystic fibrosis (CF). The CFTR gene encodes a chloride and bicarbonate channel at the apical membrane of epithelial cells. Altered ion transport of CFTR-expressing epithelia can be used to differentiate manifestations of the so-called CF basic defect. Recently, an 11p13 region has been described as a CF modifier by the North American CF Genetic Modifier Study Consortium. Selecting the epithelial-specific transcription factor EHF (ets homologous factor) as the likely candidate gene on 11p13, we have genotyped two intragenic microsatellites in EHF to replicate the 11p13 finding in the patient cohort of the European CF Twin and Sibling Study. We could observe an association of rare EHF haplotypes among homozygotes for c.1521_1523delCTT in CFTR, which exhibit a CF-untypical manifestation of the CF basic defect such as CFTR-mediated residual chloride secretion and low response to amiloride. We have reviewed transcriptome data obtained from intestinal epithelial samples of homozygotes for c.1521_1523delCTT in CFTR, which were stratified for their EHF genetic background. Transcripts that were upregulated among homozygotes for c.1521_1523delCTT in CFTR, who carry two rare EHF alleles, were enriched for genes that alter protein glycosylation and trafficking, both mechanisms being pivotal for the effective targeting of fully functional p.Phe508del-CFTR to the apical membrane of epithelial cells. We conclude that EHF modifies the CF phenotype by altering capabilities of the epithelial cell to correctly process the folding and trafficking of mutant p.Phe508del-CFTR.
Figures
Similar articles
-
An association study on contrasting cystic fibrosis endophenotypes recognizes KRT8 but not KRT18 as a modifier of cystic fibrosis disease severity and CFTR mediated residual chloride secretion.BMC Med Genet. 2011 May 6;12:62. doi: 10.1186/1471-2350-12-62. BMC Med Genet. 2011. PMID: 21548936 Free PMC article.
-
Genes that determine immunology and inflammation modify the basic defect of impaired ion conductance in cystic fibrosis epithelia.J Med Genet. 2011 Jan;48(1):24-31. doi: 10.1136/jmg.2010.080937. Epub 2010 Sep 12. J Med Genet. 2011. PMID: 20837493 Free PMC article.
-
Expression of delta F508 cystic fibrosis transmembrane conductance regulator protein and related chloride transport properties in the gallbladder epithelium from cystic fibrosis patients.Hepatology. 1999 Jun;29(6):1624-34. doi: 10.1002/hep.510290634. Hepatology. 1999. PMID: 10347100
-
Managing the underlying cause of cystic fibrosis: a future role for potentiators and correctors.Paediatr Drugs. 2013 Oct;15(5):393-402. doi: 10.1007/s40272-013-0035-3. Paediatr Drugs. 2013. PMID: 23757197 Review.
-
Genotype and phenotype in cystic fibrosis.Respiration. 2000;67(2):117-33. doi: 10.1159/000029497. Respiration. 2000. PMID: 10773783 Review.
Cited by
-
Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine.Genes (Basel). 2021 Apr 13;12(4):562. doi: 10.3390/genes12040562. Genes (Basel). 2021. PMID: 33924524 Free PMC article. Review.
-
Genotype and transcript processing of the tumour necrosis factor receptor TNFRSF1A in epithelial cells: implications for survival in cystic fibrosis.EBioMedicine. 2025 Aug;118:105848. doi: 10.1016/j.ebiom.2025.105848. Epub 2025 Jul 10. EBioMedicine. 2025. PMID: 40645008 Free PMC article.
-
Consistent Assignment of Risk and Benign Allele at rs2303153 in the CF Modifier Gene SCNN1B in Three Independent F508del-CFTR Homozygous Patient Populations.Genes (Basel). 2021 Sep 29;12(10):1554. doi: 10.3390/genes12101554. Genes (Basel). 2021. PMID: 34680949 Free PMC article.
-
The Role of Specialized Pro-Resolving Mediators in Cystic Fibrosis Airways Disease.Front Pharmacol. 2020 Sep 2;11:1290. doi: 10.3389/fphar.2020.01290. eCollection 2020. Front Pharmacol. 2020. PMID: 32982730 Free PMC article. Review.
-
Pseudomonas aeruginosa Reduces VX-809 Stimulated F508del-CFTR Chloride Secretion by Airway Epithelial Cells.PLoS One. 2015 May 27;10(5):e0127742. doi: 10.1371/journal.pone.0127742. eCollection 2015. PLoS One. 2015. PMID: 26018799 Free PMC article.
References
-
- Welsh MJ, Ramsey BW, Accurso F, Cutting GR.Cystic Fibrosisin Scriver CR (ed). The Metabolic & Molecular Bases of Inherited Disease: New York, NY; 20015121–5188.
-
- Strausbaugh SD, Davis PB. Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med. 2007;28:279–288. - PubMed
-
- Rich DP, Anderson MP, Gregory RJ, et al. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature. 1990;347:358–363. - PubMed
-
- Tang L, Fatehi M, Linsdell P. Mechanism of direct bicarbonate transport by the CFTR anion channel. J Cyst Fibros. 2009;8:115–121. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
