

Recessive TTN truncating mutations define novel forms of core myopathy with heart disease
- PMID: 24105469
- PMCID: PMC3954110
- DOI: 10.1093/hmg/ddt494
Recessive TTN truncating mutations define novel forms of core myopathy with heart disease
Abstract
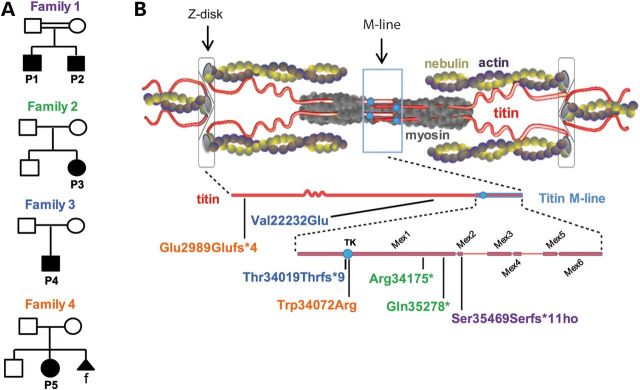
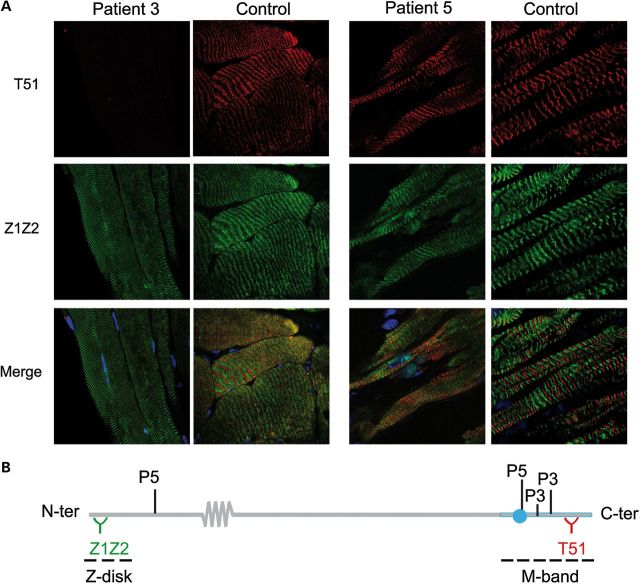
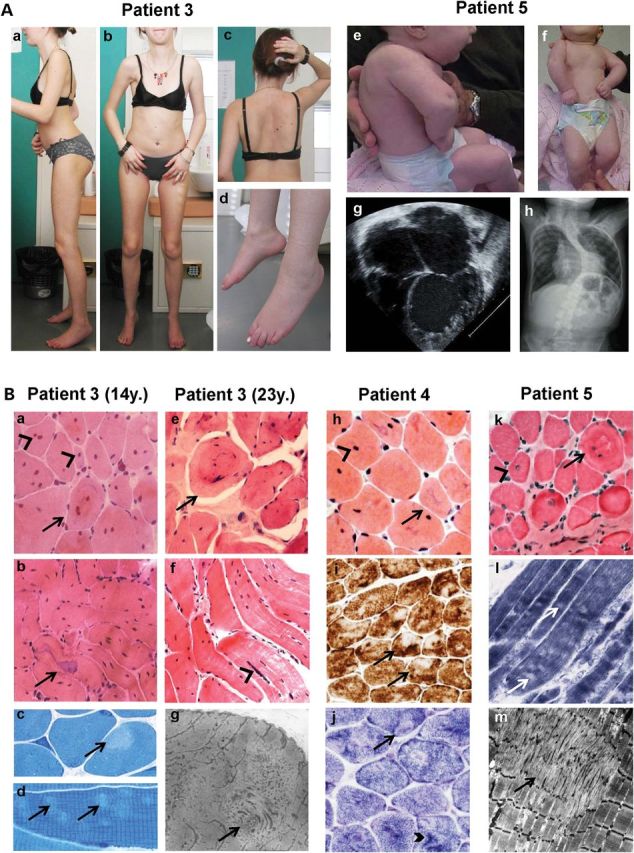
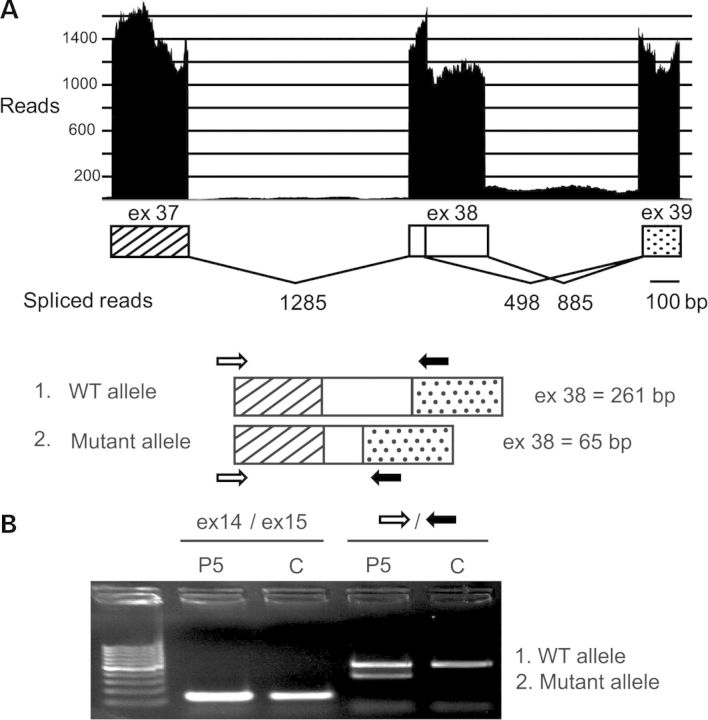
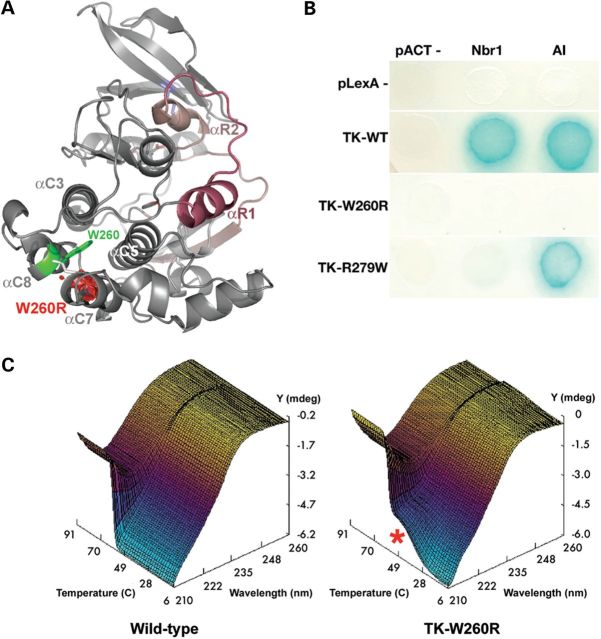
Core myopathies (CM), the main non-dystrophic myopathies in childhood, remain genetically unexplained in many cases. Heart disease is not considered part of the typical CM spectrum. No congenital heart defect has been reported, and childhood-onset cardiomyopathy has been documented in only two CM families with homozygous mutations of the TTN gene. TTN encodes titin, a giant protein of striated muscles. Recently, heterozygous TTN truncating mutations have also been reported as a major cause of dominant dilated cardiomyopathy. However, relatively few TTN mutations and phenotypes are known, and titin pathophysiological role in cardiac and skeletal muscle conditions is incompletely understood. We analyzed a series of 23 families with congenital CM and primary heart disease using TTN M-line-targeted sequencing followed in selected patients by whole-exome sequencing and functional studies. We identified seven novel homozygous or compound heterozygous TTN mutations (five in the M-line, five truncating) in 17% patients. Heterozygous parents were healthy. Phenotype analysis identified four novel titinopathies, including cardiac septal defects, left ventricular non-compaction, Emery-Dreifuss muscular dystrophy or arthrogryposis. Additionally, in vitro studies documented the first-reported absence of a functional titin kinase domain in humans, leading to a severe antenatal phenotype. We establish that CM are associated with a large range of heart conditions of which TTN mutations are a major cause, thereby expanding the TTN mutational and phenotypic spectrum. Additionally, our results suggest titin kinase implication in cardiac morphogenesis and demonstrate that heterozygous TTN truncating mutations may not manifest unless associated with a second mutation, reassessing the paradigm of their dominant expression.
Figures
References
-
- Ferreiro A., Estournet B., Chateau D., Romero N.B., Laroche C., Odent S., Toutain A., Cabello A., Fontan D., dos Santos H.G., et al. Multi-minicore disease: searching for boundaries. Phenotypical analysis of 38 cases. Ann. Neurol. 2000;48:745–757. - PubMed
-
- Ferreiro A., Monnier N., Romero N.B., Leroy J.-P., Bönnemann C., Haenggeli C.-A., Straub V., Voss W.D., Nivoche Y., Jungbluth H., et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann. Neurol. 2002;51:750–759. - PubMed
-
- Ferreiro A., Quijano-Roy S., Pichereau C., Moghadaszadeh B., Goemans N., Bönnemann C., Jungbluth H., Straub V., Villanova M., Leroy J.-P., et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am. J. Hum. Genet. 2002;71:739–749. doi:10.1086/342719. - DOI - PMC - PubMed
-
- Boyden S.E., Mahoney L.J., Kawahara G., Myers J.A., Mitsuhashi S., Estrella E.A., Duncan A.R., Dey F., DeChene E.T., Blasko-Goehringer J.M., et al. Mutations in the satellite cell gene MEGF10 cause a recessive congenital myopathy with minicores. Neurogenetics. 2012;13:115–124. doi:10.1007/s10048-012-0315-z. - DOI - PMC - PubMed
-
- Carmignac V., Salih M.A.M., Quijano-Roy S., Marchand S., Al Rayess M.M., Mukhtar M.M., Urtizberea J.A., Labeit S., Guicheney P., Leturcq F., et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann. Neurol. 2007;61:340–351. doi:10.1002/ana.21089. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
