

A single-molecule long-read survey of the human transcriptome
- PMID: 24108091
- PMCID: PMC4075632
- DOI: 10.1038/nbt.2705
A single-molecule long-read survey of the human transcriptome
Abstract
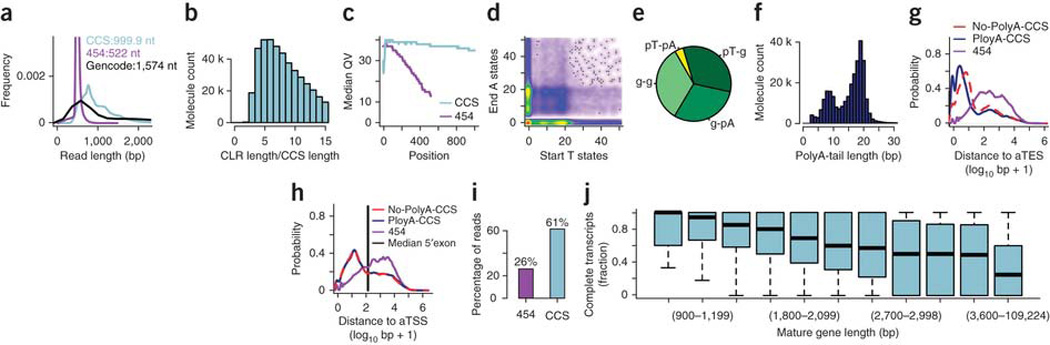
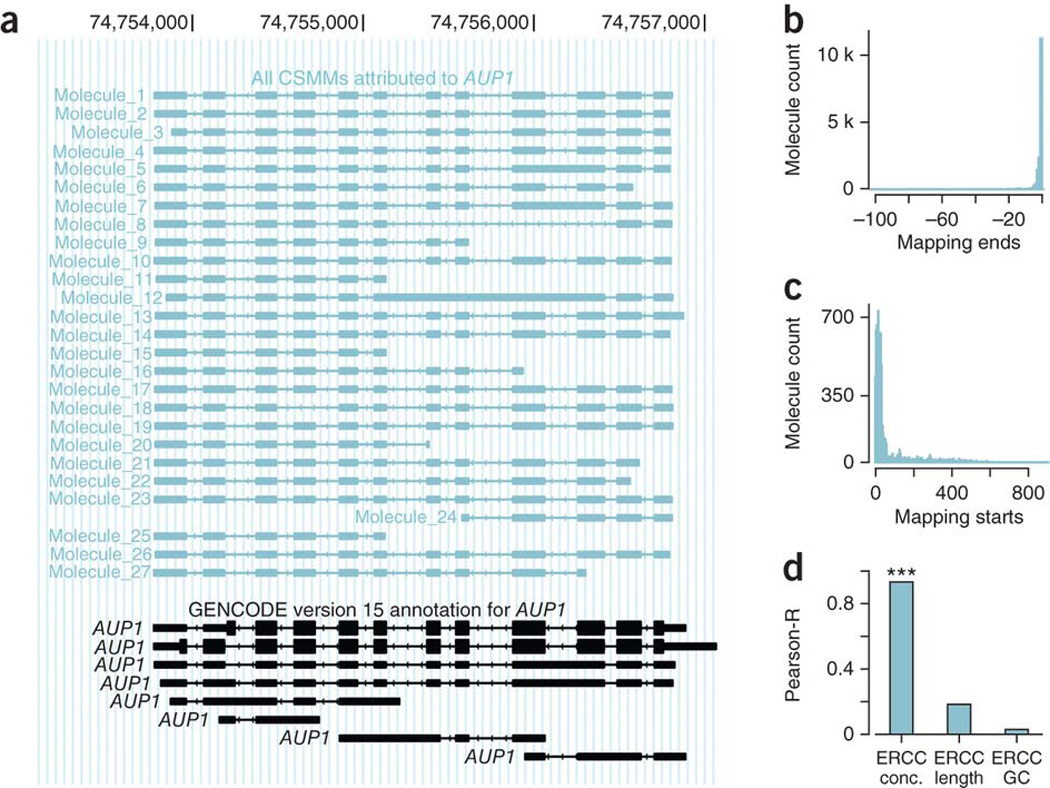
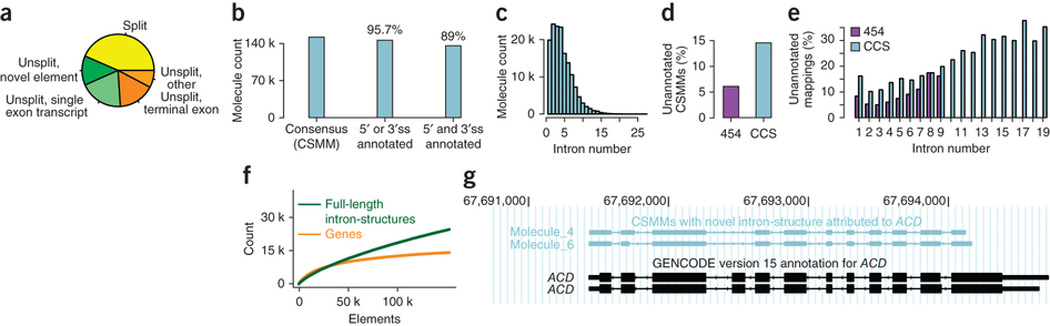
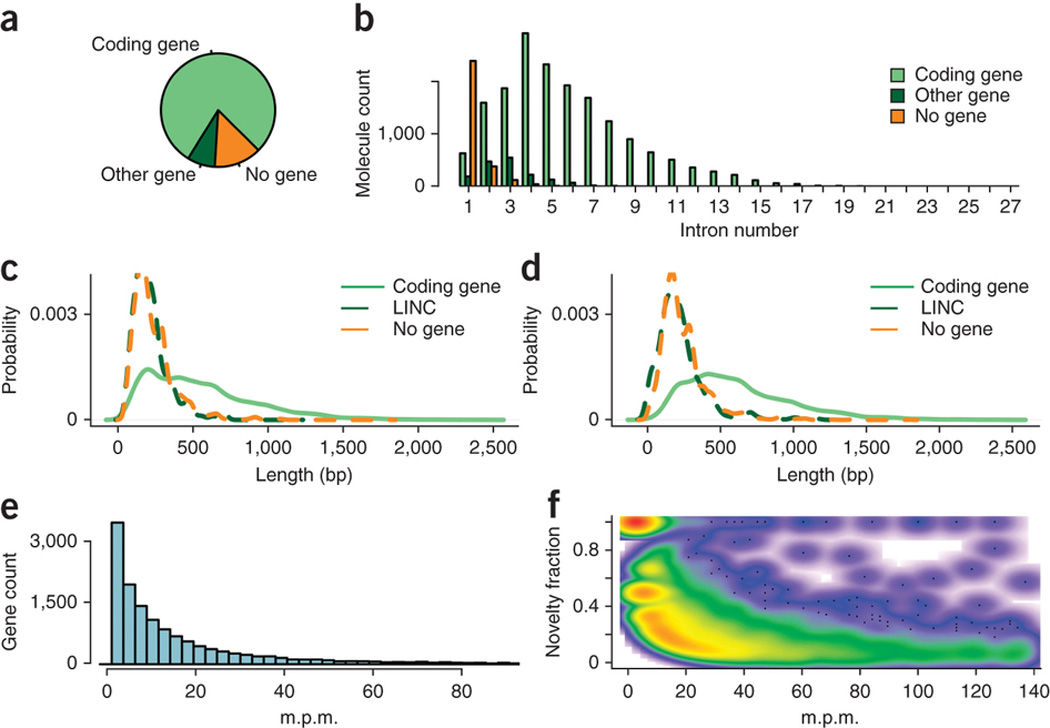
Global RNA studies have become central to understanding biological processes, but methods such as microarrays and short-read sequencing are unable to describe an entire RNA molecule from 5' to 3' end. Here we use single-molecule long-read sequencing technology from Pacific Biosciences to sequence the polyadenylated RNA complement of a pooled set of 20 human organs and tissues without the need for fragmentation or amplification. We show that full-length RNA molecules of up to 1.5 kb can readily be monitored with little sequence loss at the 5' ends. For longer RNA molecules more 5' nucleotides are missing, but complete intron structures are often preserved. In total, we identify ∼14,000 spliced GENCODE genes. High-confidence mappings are consistent with GENCODE annotations, but >10% of the alignments represent intron structures that were not previously annotated. As a group, transcripts mapping to unannotated regions have features of long, noncoding RNAs. Our results show the feasibility of deep sequencing full-length RNA from complex eukaryotic transcriptomes on a single-molecule level.
Figures
References
-
- Sultan M, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. - PubMed
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. - PubMed
-
- Wilhelm BT, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
