

Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP
- PMID: 24113144
- PMCID: PMC3900109
- DOI: 10.1093/hmg/ddt497
Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP
Abstract
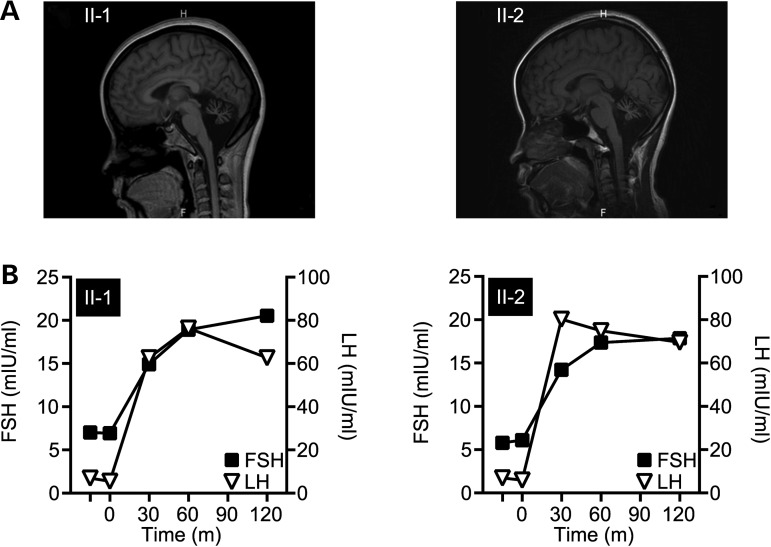
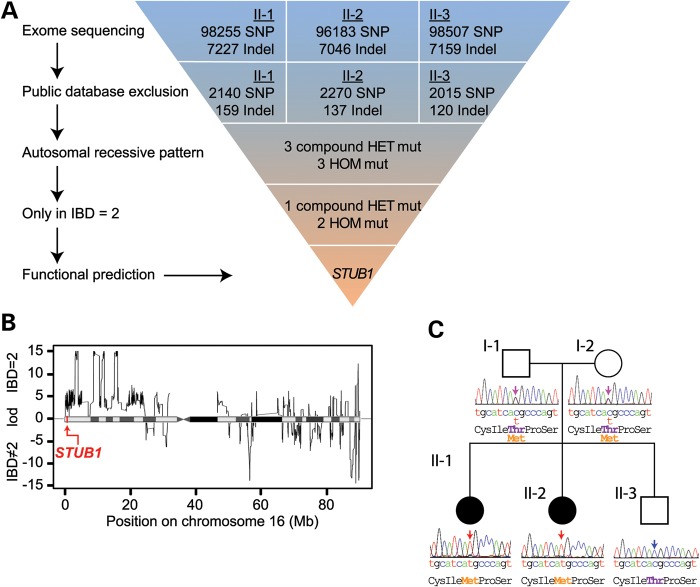
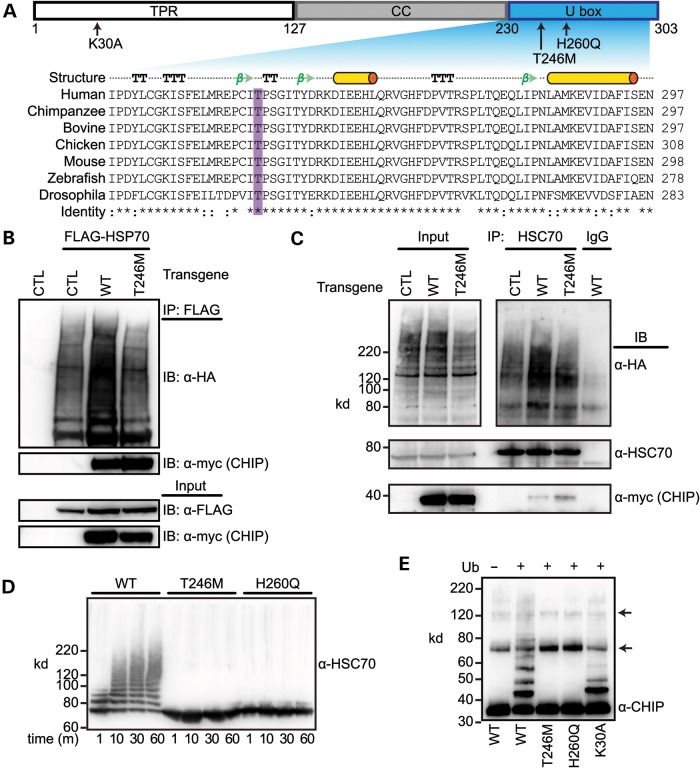
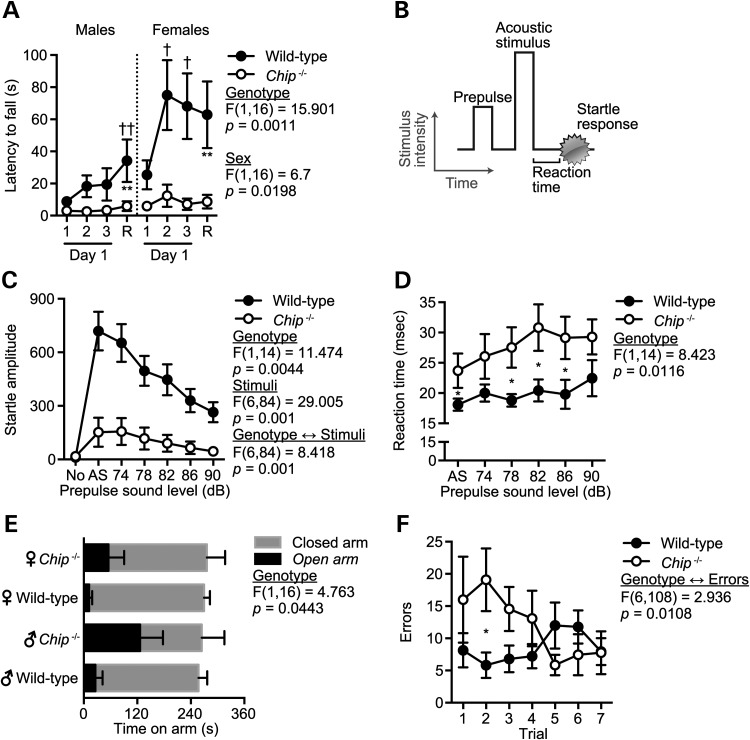
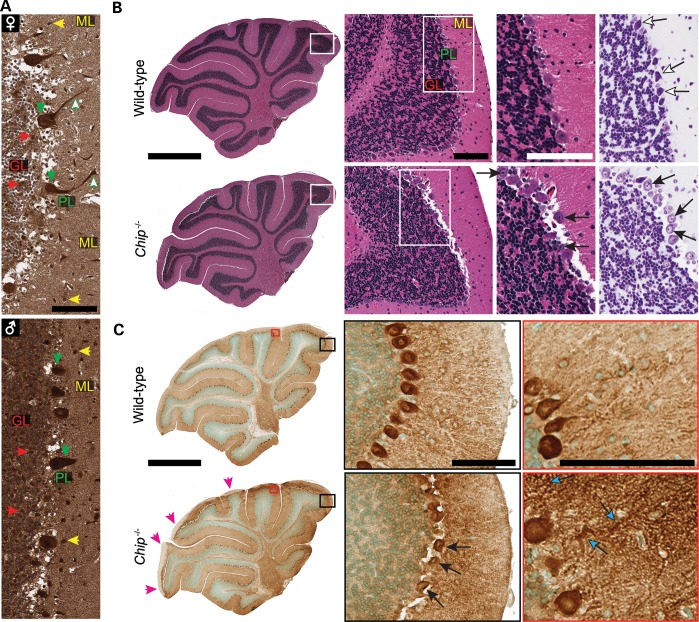
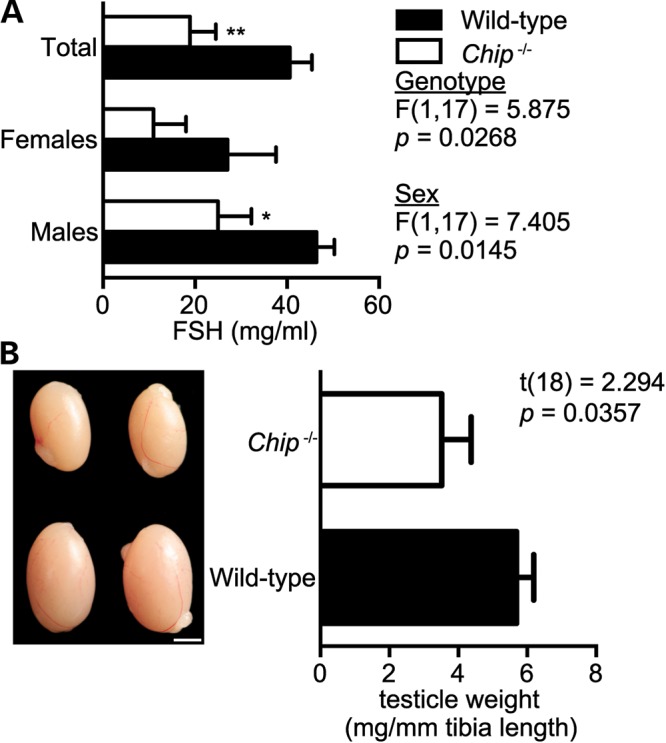
Gordon Holmes syndrome (GHS) is a rare Mendelian neurodegenerative disorder characterized by ataxia and hypogonadism. Recently, it was suggested that disordered ubiquitination underlies GHS though the discovery of exome mutations in the E3 ligase RNF216 and deubiquitinase OTUD4. We performed exome sequencing in a family with two of three siblings afflicted with ataxia and hypogonadism and identified a homozygous mutation in STUB1 (NM_005861) c.737C→T, p.Thr246Met, a gene that encodes the protein CHIP (C-terminus of HSC70-interacting protein). CHIP plays a central role in regulating protein quality control, in part through its ability to function as an E3 ligase. Loss of CHIP function has long been associated with protein misfolding and aggregation in several genetic mouse models of neurodegenerative disorders; however, a role for CHIP in human neurological disease has yet to be identified. Introduction of the Thr246Met mutation into CHIP results in a loss of ubiquitin ligase activity measured directly using recombinant proteins as well as in cell culture models. Loss of CHIP function in mice resulted in behavioral and reproductive impairments that mimic human ataxia and hypogonadism. We conclude that GHS can be caused by a loss-of-function mutation in CHIP. Our findings further highlight the role of disordered ubiquitination and protein quality control in the pathogenesis of neurodegenerative disease and demonstrate the utility of combining whole-exome sequencing with molecular analyses and animal models to define causal disease polymorphisms.
Figures
References
-
- Holmes G. A form of familial degeneration of the cerebellum. Brain. 1908;30:466–489.
-
- Connell P., Ballinger C.A., Jiang J., Wu Y., Thompson L.J., Hohfeld J., Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001;3:93–96. - PubMed
-
- Jiang J., Ballinger C.A., Wu Y., Dai Q., Cyr D.M., Hohfeld J., Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 2001;276:42938–42944. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
